
Theoretical investigation of fluorescence changes caused by methanol bridge based on ESIPT reaction*
2021-11-23 07:32XingleiZhang張星蕾LixiaZhu朱麗霞ZhengranWang王正然BifaCao曹必發QiaoZhou周悄YouLi李尤BoLi栗博HangYin尹航andYingShi石英
Chinese Physics B 2021年11期
關鍵詞:石英
Xinglei Zhang(張星蕾), Lixia Zhu(朱麗霞), Zhengran Wang(王正然), Bifa Cao(曹必發), Qiao Zhou(周悄),You Li(李尤), Bo Li(栗博), Hang Yin(尹航), and Ying Shi(石英)
Institute of Atomic and Molecular Physics,Jilin University,Changchun 130012,China
Keywords: DFT/TDDFT,fluorochromic,excited state intramolecular proton transfer,methanol bridge
1. Introduction
Excited state intramolecular proton transfer (ESIPT), as one of the most elementary processes,has been widespread in biological and chemical reactions.[1-3]ESIPT is the relocation of the proton in the excited state, generating a proton transfer tautomer.[4-6]Usually,dual fluorescence can be observed,and one is from the normal emission and another is due to the tautomeric emission with large Stokes shift.[7-9]Molecules with these unique properties have been developed and applied in laser dyes,[10]luminescent materials,[11]molecular switches,[12]and fluorescent probes.[13,14]The control and regulation of the ESIPT process still faces huge challenges.[15]It has long been known that the ESIPT reactions were sensitive to the surrounding solvent environment, which can be regulated by the interactions between solute and solvent molecules.[16-18]
Isoquinoline is a nitrogenous aromatic compound,[19]which has important application value in the fields of organic luminescence materials.[20]Isoquinoline derivatives are typical bifunctional molecule with both proton donor and acceptor groups.[21]It has always been one of the significant research directions for organic synthesis workers.[22]Gomez Pinheiroet al.synthesized a new isoquinoline derivative 3-hydroxy-4-pyridylisoquinoline (2a) mildly, with the features of solvatochromic and acidochromic.[23]In their experimental studies, it can be noted that only a single emission was observed in the tetrahydrofuran (THF) solvent, while double emission of 2a was obviously observed in the methanol(MeOH)solvent.The 2a system showed significantly sensitive emission characteristics to the solvent effects. These type of fluorosolvatochromic compounds laid a promising foundation for the development of solvent-sensitive fluorescent probes.However,the reason for this different fluorescent phenomenon is still unclear.
Herein, we investigate the proton transfer process of 2a complex in MeOH and compare it with the monomer form in THF. According to the protonation of the solvent, 2a system was divided into monomer form and hydrogen bonded complex form.[24]The geometric parameters were analyzed to characterize the nature and strength of hydrogen bonds(HBs)in 2a monomer form and complex form. The formation and fracture of HBs were analyzed by calculating infrared spectrum(IR).And the type of interaction intuitively was visually shown by using the reduced density gradient (RGD) isosurface. The electron density distribution was drawn by frontier molecular orbital. In addition,the potential energy curves were scanned to reveal the feasible transfer channels. Moreover,further explanation of the transfer mechanism of the double HBs system formed between MeOH and 2a was investigated.
2. Theoretical calculation method
All the theoretical calculation was based on the Gaussian 09 program suite.[25]The geometric of ground-state (S0) and the excited-state(S1)with B3LYP[28,29]functional and TZVP basis sets[30-32]were optimized by using the density functional theory(DFT)and time-dependent DFT(TDDFT)theory.[26,27]In order to accurately simulate the effect of MeOH on the ESIPT process of the 2a system, the explicit solvent model was selected to calculate the 2a-MeOH complex, which was coordinated by a MeOH molecule via two HBs.[33-36]The effect of protonation of solvent on 2a system was simulated based on the polarizable continuum model (PCM) using the integral equation formalism variant(IEFPCM).[37,38]In addition, to clarify the characteristic of intra- and inter-molecular HBs, the non-covalent interactions (NCI) analysis was executed by Multiwfn program.[39,40]The potential energy curves were scanned along the stretching direction at a step of 0.1 ?A.
3. Results and discussion
3.1. Optimized geometric structures
The optimized geometry structures of 2a and 2a-MeOH complex(enol and keto forms)in S0and S1states are shown in Fig. 1. For 2a monomer form in THF, the intramolecular HB can form between the proton donor (-OH) and the proton acceptor(-N-)located in the aryl isoquinoline.[41]While in MeOH solvent, the nitrogen and hydroxyl groups on 2a molecule coordinate with an explicit solvent molecule forming a complex with the formation of two intermolecular HBs.We have labelled the specific atoms associated with proton transfer to facilitate the observation of structural parameters.In addition,the geometric parameters of enol form are shown in Table 1. For 2a-E form, uniquely, the intermolecular HB H1···N1, where proton transfer can occur, increased from 2.113 ?A to 2.198 ?A,and the bond length of O1-H1decreased from 0.977 ?A to 0.973 ?A,upon excitation to the S1state. The variation of parameters demonstrated that the proton on the hydroxyl group is less likely to break away,and it would be more stable than S0state. Further,the prolonged intramolecular HB is not conducive to the ESIPT process.
In the case of 2a-MeOH-E form, the coordination intermolecular HBs are labeled as HB1(O2-H2···O3) and HB2(O3-H3···N2), which form a proton-bridge.[42]Comparing with the S0state, the bond length of O2-H2and O3-H3are both increased from 0.989 ?A to 1.005 ?A and 0.979 ?A to 0.982 ?A,respectively. The H2and H3atoms show a tendency to move away from the donor. The intermolecular HB1and HB2are shortened from 1.761 ?A to 1.665 ?A and 1.981 ?A to 1.954 ?A, respectively. The narrow of HB1and HB2in the S1state indicate that the interaction between 2a and MeOH is intensified which may stimulate the ESIPT process.[43]As a result,by comparing the distance between the hydrogen atom and the donor in the monomer and complex, it can be known that the proton is more likely to depart from hydroxyl in 2a-MeOH complex, and therefore undergo ESIPT process.[44]Furthermore, in 2a-MeOH keto form, two new intermolecular HBs emerge in the S1state,labeled as HB3(O3-H2···O2)and HB4(N2-H3···O3). The appearance of tautomer proves the occurrence of ESIPT process through MeOH-bridge.

Fig. 1. The optimized geometries of 2a and 2a-MeOH calculated at the B3LYP/TZVP theoretical level.
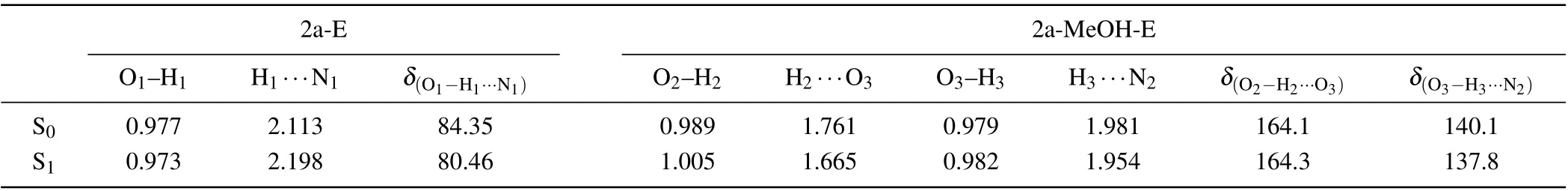
Table 1. Bond lengths(?A)and angles(°)associated with hydrogen bonding of 2a monomer and 2a-MeOH in the S0 and S1 states.
3.2. Infrared vibrational spectra
Monitoring the movement of O-H group stretching vibration in IR spectrum can characterize the change of the corresponding HB intensity. The calculation results of the IR spectra of the 2a monomer and 2a-MeOH complex in the S0state and the S1state are shown in Fig. 2. For the monomer, the stretching vibration frequency of O1-H1move in the direction of greater wavenumber shows blue-shift. It is difficult for the detachment of proton as the stretching vibration requires more energy. Then after the formation of 2a-MeOH complex,the O2-H2group in the ground state shows a significant red shift to 3296 cm?1from 3688 cm?1in 2a monomer. And the red-shift further moves to 3003 cm?1when the complex is excited to the S1state. The movement of stretching vibration frequency peaks indicates that the vibration energy required for the O-H group is reduced,in which the proton tends to be transferred away from the donor. The vibration peak corresponding to the O3-H3group in MeOH solvent has the shift from the location of 3520 cm?1in the S0state to 3465 cm?1in the S1state. Therefore,the HB1and HB2in the excited state have similar movement toward the smaller wavenumber.[45]It means that the bond energy is strengthened, which is consistent with the conclusions drawn from the previous molecular structure parameters. Compared with the monomer,the complex exhibits proton movement reaction,and it becomes easier after being excited to the S1state. It can be concluded that the bridging by MeOH molecular has significant facilitate effects on the ESIPT process in 2a system.

Fig.2. The calculated IR spectra of 2a monomer(a)and 2a-MeOH complex(b)in the S0 and S1 states.
3.3. Electronic spectra and frontier molecular orbitals
The electronic transition properties in the first two excited states of 2a monomer and 2a complex are presented in Table 2. The vertical excitation energy of 2a monomer and 2a complex are in good agreement with the experimental absorption in THF (355 nm) and MeOH (354 nm),[23]respectively.It verifies that the function and base set used in the calculation are appropriate. Moreover, the transitions from S0to S1correspond to the maximum oscillator strength for both monomer and complex structures.Therefore,only the S0→S1transition is investigated in this paper.
Front molecular orbitals are used to describe the charge redistribution and transfer characteristics of excited states as depicted in the Fig. 3. The transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital(LUMO)corresponds to a distinctπ-π*type feature,for both monomer and complex form. The charge distribution shifts from hydroxyl to N1atom in these two systems,makes the hydroxyl more acidic and the isoquinoline becomes more basic.[46]These acid-base changes can promote the occurrence of proton transfer process. It is worth noting that there is almost no charge density on the MeOH molecule,indicating that the methanol molecule is still in the ground state while only the 2a molecule was electronically excited.[47]Furthermore, by comparing the LUMO in the two systems, it can be found that the charge distribution on hydroxyl are significantly reduced in the 2a-MeOH complex than that in the 2a monomer. The pyridine ring is twisted, and the change of structure leads to the different optical properties.[48]This also proves that the addition of MeOH molecule makes the 2a molecule more prone to proton transfer.

Fig.3. The HOMO and LUMO orbitals of 2a monomer and 2a-MeOH complex structures.
3.4. Non-covalent interactions analysis
The non-covalent interactions (NCI) provide conditions for identifying the interaction and understanding the corresponding intensities.[49]The electron densitiesρ(r) analysis and the reduced density gradient(RDG)isosurfaces visualization is one of the methods.[50]In addition, the second largest eigenvalue of Hessian matrix of electron densityλ2(r) was also demanded. The interaction exhibits attractive ifλ2(r) is less than zero and displays repulsive ifλ2(r)exceeds zero.The relevant functions are shown as below:
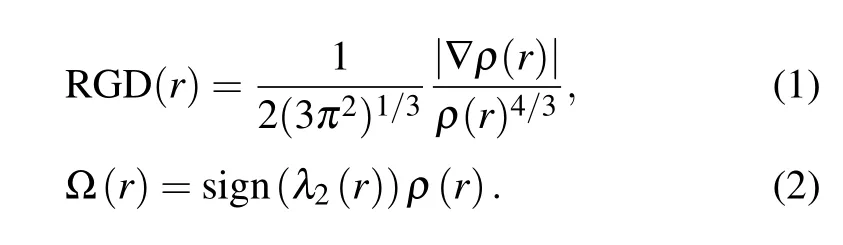
The scatter diagrams of the RDG(r) value versus the Ω(r)value of 2a and 2a-MeOH are mapped out in Fig. 4. The value of the function represents in different colors according to the color gradient bar below, and clearly presents the different NCI types. Therefore, we can not only know the occurrence area of weak interaction,but also can distinguish the type of interaction intuitively. For the 2a monomer, there is a red disk between oxygen and hydrogen atoms corresponding to the positive value about 0.023 in the scattergram. After being excited to the S1state,the interaction corresponding to the peak position of scattered points is still strong repulsion. It clarifies why it is difficult for the monomer form to undergo a proton transfer process. However,for 2a-MeOH complex,the elliptic plates at the junction of 2a and MeOH show blue color which represent strong hydrogen bond interactions. It can be seen that the blue color is deep upon excitation to the S1state.In addition, the dark blue and light blue pieces correspond to HB1and HB2, respectively. The value of HB1spike shifted from?0.041 in the S0state to?0.052 in the S1state, and the value of HB2spike in the shifted from?0.03 to?0.032 in the S1state compared with the S0state. The color difference and spikes shift of the HBs describe the enhancement of intramolecular interactions upon excited into the S1state.And these results,once again,emphasize that the involving of MeOH effectively promote the ESIPT process and keep highly consistent with the above discussions.

Table 2. Calculated electronic excitation energies(nm)and corresponding oscillator strengths(OS)of 2a monomer and 2a-MeOH complex.
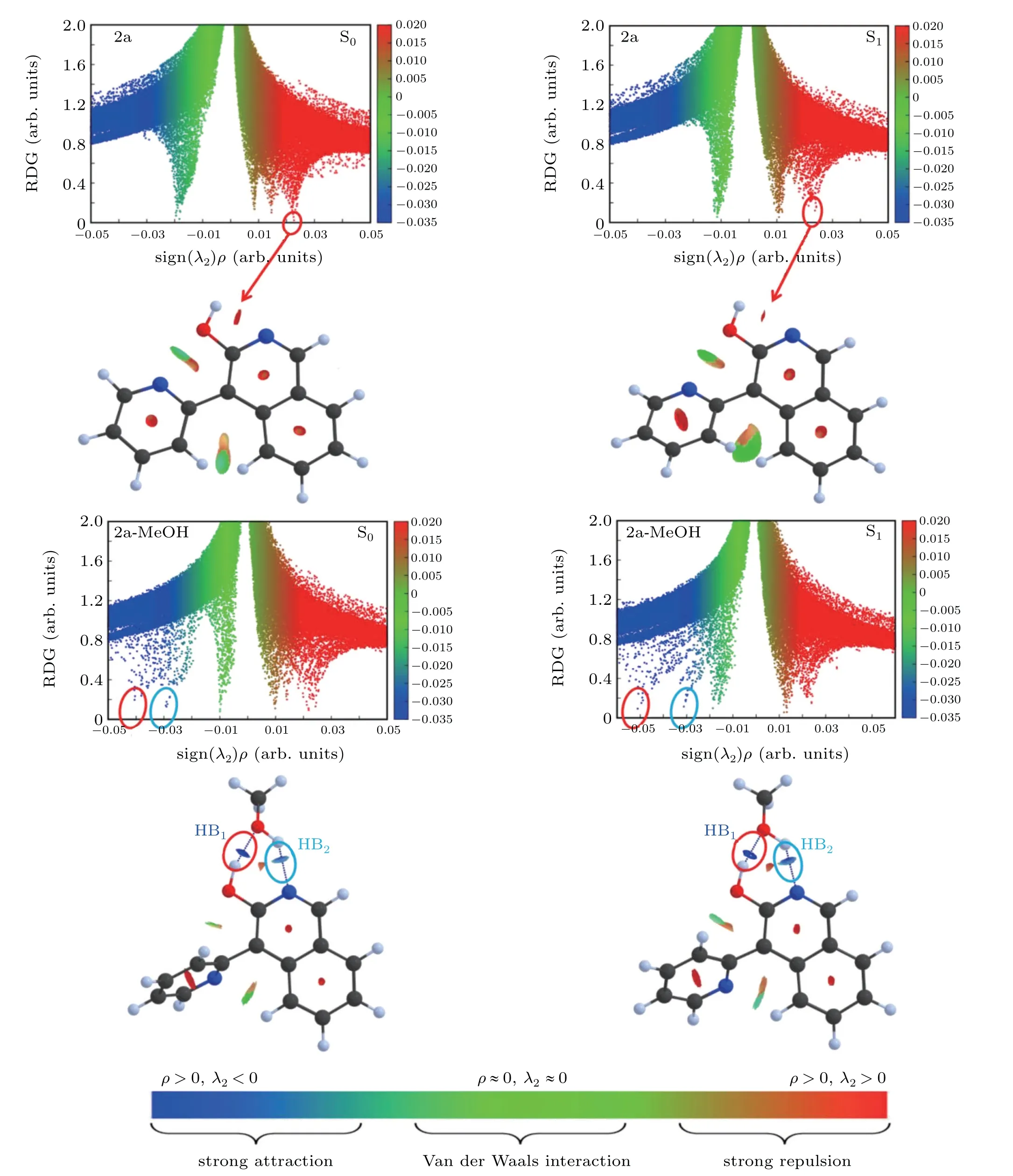
Fig.4. The RDG scatter diagrams and corresponding isosurfaces of 2a and 2a-MeOH for enol forms at S0 and S1 states.
3.5. Potential energy curves
The protic solvent effect on the ESIPT process can be revealed by constructing the potential energy curve. The molecular energy of S0state and S1state in two solvents was calculated with the variation of hydrogen bond distance, as shown in Fig. 5. The 2a monomer form has one pathway from proton donor (O-H) to proton acceptor (-N-) since aprotic solvent THF can neither provide protons nor break intramolecular HBs. The energy along the O-H bond in 2a shows that the barrier was 31.41 kcal/mol in S0state and reduced to 25.01 kcal/mol in S1state. Therefore, the proton transfer reaction is difficult to occur for both S0and S1states.[51]
As a typical solvent with both proton donor and receptor, enables 2a to undergo ESIPT process assisted by MeOH molecule. The potential transfer channels of 2a-MeOH were considered to have three types which were depicted alongside the energy curves.[52]The stepwise mechanism is discussed firstly.The proton transferred from solute 2a to solvent MeOH(type 1)or from solvent MeOH to solute 2a(type 2),each proton transfer process occurs sequentially. The other pathway follows concerted mechanism in which proton transfer process occurs simultaneously along HB1and HB2(type 3). For type 1-3 pathways, the energy barriers are 11.47, 12.19, and 11.81 kcal/mol in the S0state,respectively.The relatively high energy barrier makes proton transfer process hardly occur in the S0state.[53]While in the S1state,the barrier was reduced to 4.8, 6.83, and 6.42 kcal/mol, respectively. Obviously, the type 1 stepwise mechanism seems to be an easier barrier for the proton transfer reaction. The H2atom on the isoquinoline departs in the first step,transfers to the MeOH along the intermolecular HB1and then following the H3atom moves to the O2atom via the intermolecular HB2. As a consequence,with the aid of a bridge composed by single MeOH molecule, the proton transfer energy barrier was effectively reduced. And the ESIPT process was successfully implemented through the sequential type reaction channel.
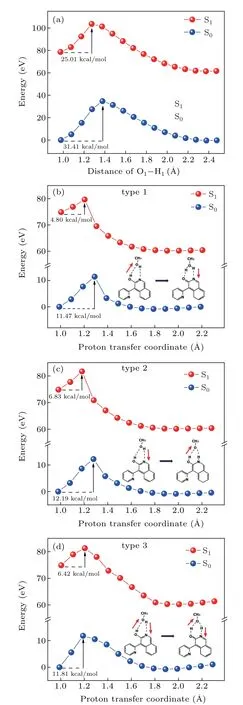
Fig. 5. The potential energy curves of 2a monomer (a) and three potential pathways of 2a-MeOH complex,(b)type 1,(c)type 2,(d)type 3,at S0 and S1 states.
3.6. Absorption and emission spectra
The corresponding absorption and fluorescence spectra of 2a and 2a-MeOH are shown in Fig.6. The simulated computation of absorption peaks at 359 nm and 342 nm stay closely aligned with the experimental data(355 nm and 354 nm),[23]respectively. For 2a monomer, two emission peaks are obtained by optimized structure which correspond to the 2a-E and 2a-K states. As a matter of fact, only one emission was detected in experiment. These are consistent with the previous scanning result of the potential energy curves and the proton cannot transfer according to the high energy barrier. In addition,the computation peak of the 2a monomer in the E state at 407 nm is in accord with the single emission at 401 nm. As a result, the single fluorescence peak in THF solvent is ascribe to the 2a-E form.

Fig.6.The calculated absorption and fluorescence spectra of 2a monomer(a)and 2a-MeOH complex(b)forms(data in brackets are experimental values).
While in the MeOH solvent, after photoexcitation, there are two emission peaks at 423 nm and 516 nm which separately coincident with the experimental data 398 nm and 519 nm.[23]According to the scanned potential energy curve,as the solvent molecular involved in the reaction, the 2a-MeOH complex possesses an ESIPT process in sequential way after excitation. Thus, the first fluorescence meets with 2a-MeOH-E*(enol from in S1state) and the dual fluorescence is derived from the tautomer 2a-MeOH-K*(keto from in S1state). In conclusion, the bridging effect of MeOH promotes the occurrence of dual emission of 2a system.
4. Conclusion
In summary, we have theoretically investigated the fluorescence changes induced by methanol bridge based on ESIPT reaction in 2a system. After bridging by MeOH molecule,the OH bond stretching vibration frequency spike has a blueshift in 2a monomer and it turns into a red-shift in 2a complex upon excitation. The charge distribution on the hydroxy of 2a dramatically decreased, indicating that proton can be easily deviated. And RDG analysis shows that the participation of MeOH changed interaction from strong repulsion into strong mutual attraction. These results prove that the bridging of MeOH is completely indispensable for the ESIPT process. Upon photo-excitation, the barrier of proton transfer is 25.01 kcal/mol for 2a monomer while it drops to 4.80 kcal/mol for 2a-MeOH complex. The significantly decrease of the ESIPT barrier will contribute to the ESIPT process effectively.Concurrently,the mechanism of ESIPT in 2a-MeOH has been carefully clarified that the transfer began from 2a molecular via stepwise reaction channel.As a consequence,the active involvement of the MeOH bridges a viable pathway for the ESIPT process and results in fluorochromic phenomenon.These unique characteristics provide valuable guidance for controlling ESIPT processes and designing and synthesizing similar molecules to improve the performance of luminescent materials in the future.
猜你喜歡
Chinese Physics B(2022年4期)2022-04-12
礦產保護與利用(2022年5期)2022-03-28
中國非金屬礦工業導刊(2020年5期)2020-11-08
科學導報(2020年50期)2020-09-09
礦冶工程(2020年2期)2020-05-24
沉積與特提斯地質(2018年2期)2018-11-22
電子制作(2018年14期)2018-08-21
錄井工程(2017年3期)2018-01-22
中國繼續醫學教育(2015年1期)2016-01-06
鐵道科學與工程學報(2015年5期)2015-12-24

- Chinese Physics B的其它文章
- Numerical investigation on threading dislocation bending with InAs/GaAs quantum dots*
- Connes distance of 2D harmonic oscillators in quantum phase space*
- Effect of external electric field on the terahertz transmission characteristics of electrolyte solutions*
- Classical-field description of Bose-Einstein condensation of parallel light in a nonlinear optical cavity*
- Dense coding capacity in correlated noisy channels with weak measurement*
- Probability density and oscillating period of magnetopolaron in parabolic quantum dot in the presence of Rashba effect and temperature*