
New flavanone-monoterpene hybrids as α-glucosidase inhibitors from the root bark of Morus alba
2022-12-06 10:42LinLinTianHuaZhang
TMR Modern Herbal Medicine 2022年4期
Lin-Lin Tian, Hua Zhang,*
1 School of Biological Science and Technology, University of Jinan, Jinan 250022, China.
Abstract
Objective The Morus alba root bark is a well-known Chinese herbal medicine called Sang-Bai-Pi and has often been used to relieve the hyperglycemic symptom of diabetes patients. The current work aims to further explore its bioactive constituents with α-glucosidase inhibitory activity for the potential treatment of diabetes.
Methods A combination of different separating techniques including routine column chromatograph and HPLC especially on chiral columns were applied for the isolation of target molecules, while comprehensive spectroscopic experiments comprising MS, NMR, ECD, etc. were carried out to complete the structural assignment. The anti-hyperglycemic property of the isolates was evaluated by an in vitro α-glucosidase inhibitory bioassay.
Results Two pairs of new flavanone-monoterpene hybrid enantiomers were isolated and identified,and an interesting phenomenon of mutual transformation between these cometabolites were detected,which resulted in their regio-isomerization and enantiomerization. The bioassay results revealed remarkable α-glucosidase inhibitory activity for these fascinating molecules.
Conclusions The Morus alba root bark is a rich source of bioactive flavonoid derivatives and deserves further investigations to develop new potential chemotherapies for diabetes control and treatment.
Keywords Morus alba; Flavanone, Regio-isomerization; Enantiomerization; α-Glucosidase inhibition
Highlights
1. Two pairs of new flavanone-monoterpene hybrid enantiomers were separated and structurally characterized fromMorus albaroot bark;
2. An interesting transformation between these cometabolites leading to regio-isomerization and enantiomerization was observed;
3. The equilibrium mixtures showed significant inhibition against the diabetes target α-glucosidase.
Introduction
Morus albaL. is likely the most famous plant species of family Moraceae owing to its tremendous economic values with both dietary and medicinal benefits [1].Its fruits, also known as mulberry, have been serving as a food for thousands of years in China and its tender shoots have also been developed into a vegetable dish by local residents of Huzhou city. Meanwhile, the twigs and root barks ofM. albahave been listed by the National Health Commission of China in a catalogue of medicinal species that can be added into functional foods. Therefore, the investigations onM. albahave always been a front-line topic in the scientific community as summarized in recent years’ review articles [2?5].
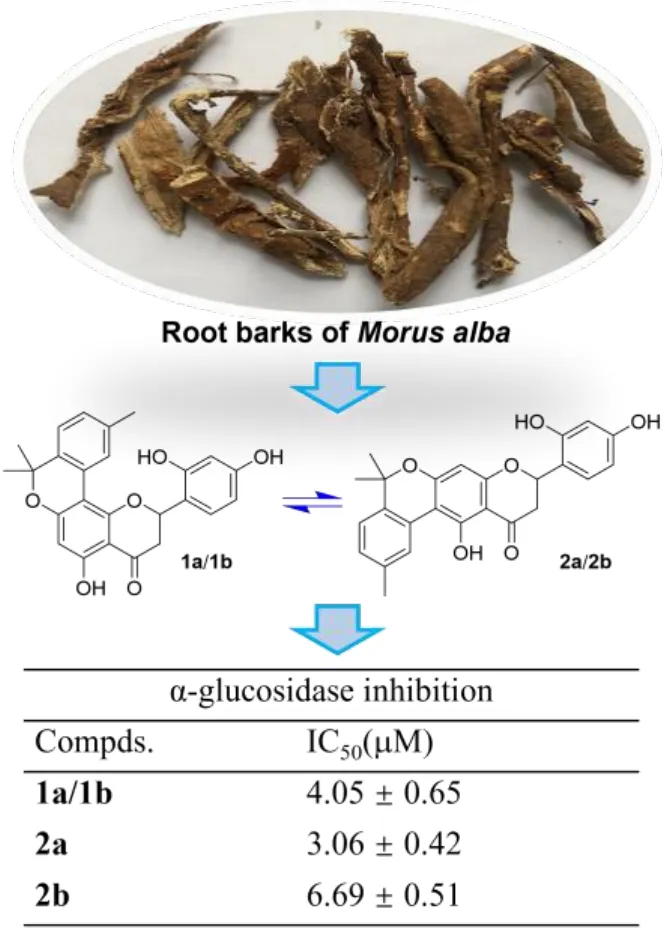
The root barks ofM. albaare a well-known species collected by the National Pharmacopeia of China as one of the commonly used herbal medicines, and they have been used to treat edema, cough, dyspnea, etc. in traditional Chinese medicine [6]. Although the study on the root barks of the title plant can be tracked back to a half-century ago [7], related phytochemical and pharmacological researches have still been very active in recent years. These investigations have revealed that theM. albaroot barks represent a very rich source of bioactive phenolic compounds especially prenylated flavonoids and benzofuran derivatives that have shown a variety of biological activities comprising anti-diabetic [8, 9],anti-inflammatory [10?12], anti-tumor [13, 14], anti-neurodegenerative [15, 16],and antiviral properties [12, 17]. As a continuation of our previous work for antihyperglycemic agents from small molecule natural products in medicinal plants[18?20], we have recently carried out an intensive study on the bioactive constituents from the root barks ofM. alba, leading to the discovery of two pairs of flavanone-monoterpene hybrid enantiomers (Figure 1). The isolation, structure identification and α-glucosidase inhibitory evaluation of these compounds will be presented here.

Figure 1. Chemical structures of compounds from the root bark of Morus alba.
Materials and Methods
General instrumentation and reagents
[α]Dvalues were measured on a Rudolph VI polarimeter (Rudolph Research Analytical, Hackettstown, USA) using a 10 cm length cell. UV and ECD spectra were acquired on a Chirascan Spectrometer (Applied Photophysics Ltd.,Leatherhead, UK). NMR experiments were performed on a Bruker Avance DRX600 spectrometer (Bruker BioSpin AG, F?llanden, Switzerland) and the residual peaks of deuterated solvents (methanol-d4: δC49.00 and δH3.31 ppm;acetone-d6: δC29.84, 206.26 and δH2.05) were used as internal references. HRESIMS spectra were recorded on an Agilent 6545 Q-TOF mass spectrometer(Agilent Technologies Inc., Waldbronn, Germany). D-101 macroporous resin(Sinopharm Chemical Reagent Co. Ltd., Shanghai, China), reversed-phase (RP)C18(Merck KGaA, Darmstadt, Germany) and silica gel (300?400 mesh, Qingdao Marine Chemical Co. Ltd., Qingdao, China) were used for routine column chromatography (CC). Agilent SB-C18column (5 μm, 9.4×250 mm, Agilent Technologies Inc., Santa Clara, USA) and Daicel AD-H chiral column (5 μm,4.6×250 mm, Daicel Chiral Technologies Co. Ltd., Shanghai, China) were used for HPLC analyses and separations. Thin-layer chromatography (TLC) was conducted with pre-coated silica gel GF254plates (Qingdao Marine Chemical Co.Ltd., Qingdao, China). Analytical and HPLC grade solvents were acquired from Tianjin Fuyu Fine Chemical Co. Ltd. (Tianjin, China) and Oceanpak Alexative Chemical Ltd. (Goteborg, Sweden), respectively.
Plant materials
The roots ofMorus albaL. (family Moraceae) were collected in August 2016 in Mount Kunyu, Shandong province, China, and the plant materials were identified by Prof. Guo-hua Ye from Shandong College of Traditional Chinese Medicine.The voucher specimen has been deposited in the author’s institution with an accession number npmc-025.
Extraction and isolation
The barks ofM. albaroots (30 kg) were peeled, dried and milled, and the powder was extracted with 95% EtOH (50 L) at r.t. for three times (once a week). The extracting solution was condensed under reduced pressure and the residue (3.6 kg)was re-suspended in 5.0 L water and partitioned with ethyl acetate (3.0 L × 3). The organic phase was solvent-removed (1.5 kg) and then fractionated by a macroporous resin CC with 30% (30 L), 50% (50 L), 80% (80 L) and 95% (50 L)EtOH-H2O successively. Each eluting component was concentrated under reduced pressure on a large-scale rotary evaporator. The 80% EtOH-H2O eluent (600 g)was separated by silica gel CC (petroleum ether-EtOAc, 20:1 to 1:2) to give 14 fractions A to N, and fraction K (4.0 g) was subsequently processed by a RP-C18column (50%?100% MeOH-H2O) to obtain eight further subfractions (K1 to K8).Subfraction K6 was eluted with CH2Cl2-MeOH (1000:1 to 100:1) on a silica gel column to afford seven new eluents (K6-1 to K6-7), and then the K6-6 eluent was purified by semi-preparative HPLC on an Agilent SB-C18column eluted with 70% MeCN-H2O (3.0 mL/min) to yield a mixture containing compounds 1a/1b and 2a/2b (2.3 mg,tR= 18.0 min). This mixture was further fractionated by a Daicel AD-H column eluted with 12% isopropanol-n-hexane (1.0 mL/min) to give compounds 1a (0.2 mg,tR= 9.5 min), 1b (0.4 mg,tR= 10.5 min), 2a (0.5 mg,tR= 16.0 min) and 2b (0.8 mg,tR= 19.0 min).
Compounds 1a/1b and 2a/2b: Yellow amorphous powder; [α]27D~0; UV(MeOH)λmax(logε) 277 (4.72) nm;1H and13C NMR data, see Table 1; (+)-ESIMSm/z419.3 [M + H]+; (+)-HR-ESIMSm/z419.1487 [M + H]+(calcd for C25H22O6, 419.1495).
α-Glucosidase inhibitory assay.
Thein vitroinhibition against α-glucosidase of compounds 1a/1b, 2a and 2b was screened according to the protocol documented previously [21]. The testing concentration of α-glucosidase was set as 0.4 U/mL. The inhibitory activity of the three samples was first tested at 100 μM, and all samples showing >50% inhibition ratio would be sent for further IC50measurements. The positive control acarbose(Aladdin, Shanghai, China) showed an IC50of 359.10 ± 2.39 μM in the present work.
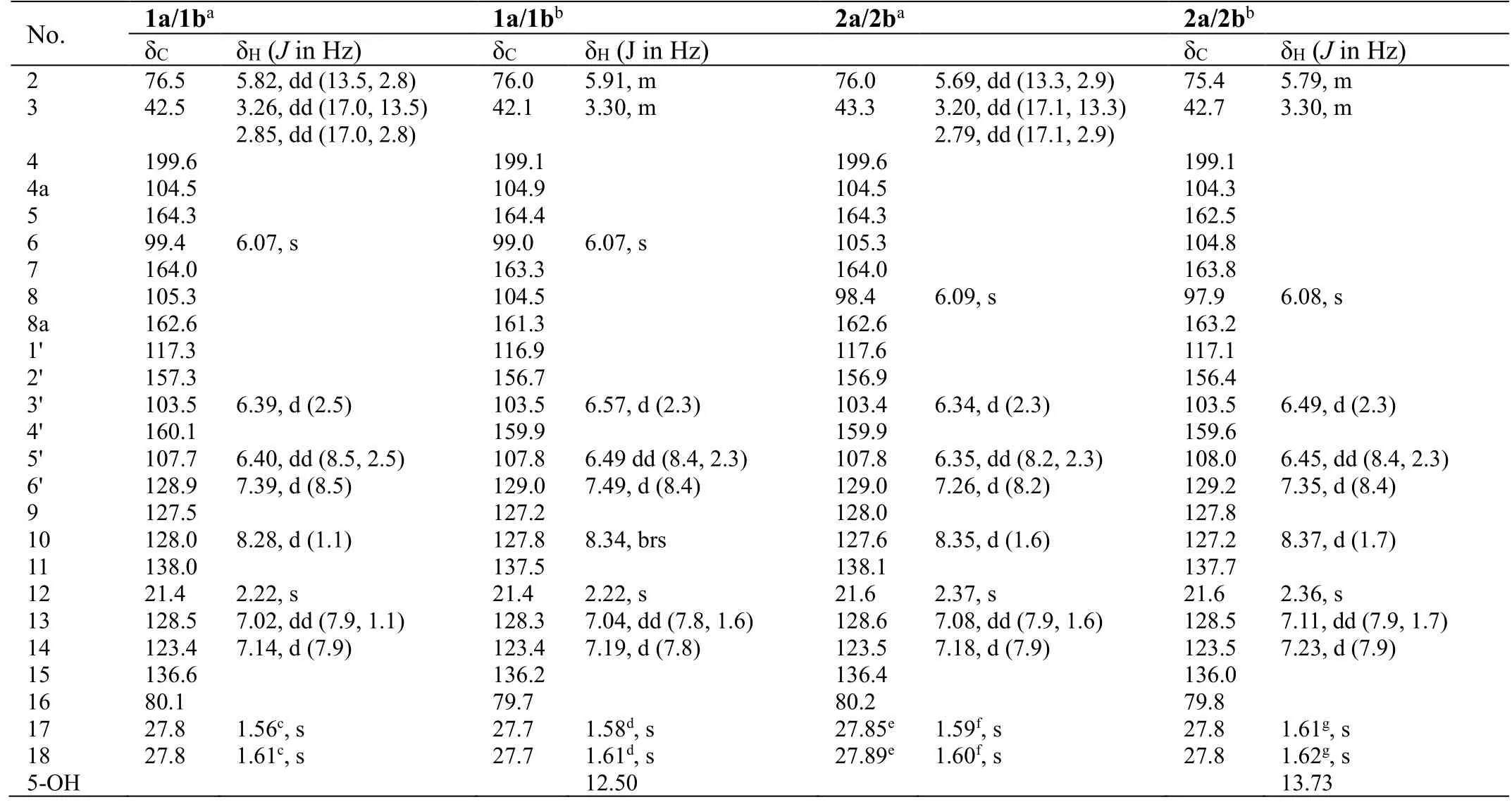
Table 1. NMR data for compounds 1a/1b and 2a/2b
Results and Discussion
Compounds 1a/1b and 2a/2b were first obtained via routine HPLC separation as yellow amorphous powder, with the molecular formula C25H22O6determined by HR-ESIMS analysis atm/z419.1487 ([M + H]+, calcd for 419.1495). The1H and13C NMR spectra of this sample in both CD3OD and CD3COCD3(Supplemental Figures S1, S2, S7 and S8) exhibited two sets of signals indicating the presence of two different molecules. However, subsequent fractionation of this mixture on several other normal HPLC columns did not gain further separation. A Daicel ADH chiral column was then used to check for further separation, which unexpectedly revealed four peaks (1a, 1b, 2a and 2b) in the HPLC chromatogram (Figure 2).The1H NMR spectra of the two major peaks (2a and 2b) were then measured in CD3OD and CD3COCD3, respectively, and the spectra were compared to those of the original mixture sample (Supplemental Figure S20), which demonstrated that both of them had identical1H NMR data with those of the major component, while those of the one (1b) from the two minor peaks were also measured in the two deuterium solvents and proved to be identical with those of the minor component in the mixture. These observations indicated that the two sets of signals in the initial NMR spectra corresponded to two pairs of enantiomers, namely 1a/1b and 2a/2b (Figure 2).

Figure 2. Chiral HPLC separation for 1a, 1b, 2a and 2b.
It was confusing that the two pairs of enantiomers 1a/1b and 2a/2b were well separated on the chiral column (Figure 2) but their NMR spectra after chiral HPLC purification still showed signals of a mixture, which was comparable to the original sample. This phenomenon implied a possible transformation between the two pairs of molecules, and their nearly flat ECD curves (Supplemental Figure S21) and small or close-to-zero [α]Dvalues (Supplemental Figures S22?S25)further suggested the likely presence of enantiomerization during their mutual isomerization process, which resulted in an equilibration among them. Therefore,the structure characterization at this stage was continued via interpretation of the spectroscopic data of the mixture.
Analyses of the NMR data (in CD3OD, Table 1) for the minor components 1a/1b revealed the presence of a carbonyl (δC199.6), an oxygenated methine [δC76.5; δH5.82 (dd,J= 13.5, 2.8 Hz)] and a methylene [δC42.5; δH3.26 (dd,J=17.0, 13.5 Hz), 2.85 (dd,J= 17.0, 2.8 Hz)] groups, which were characteristic for the C-4, CH-2 and CH2-3 of a flavanone like sanggenol L (Figure 1) [22]. More signals for a 1,2,4-trisubstituted benzene at δH7.39 (d,J= 8.5 Hz), 6.40 (dd,J=8.5, 2.5 Hz) and 6.39 (d,J= 2.5 Hz), and at δC160.1, 157.3, 128.9, 117.3, 107.7 and 103.5, as well as for a penta-substituted benzene at δH6.07 (s) and at δC164.3,164.0, 162.6, 105.3, 104.5 and 99.4, were also observed, and the two fragments corresponded to the B and A rings of the flavanone core, respectively, as in sanggenol L [22]. The major difference between the two analogues was attributed to that the monoterpenyl forming a pyran unit with ring A in sanggenol L rearranged to form a benzopyran unit with ring A in 1a/1b, which was evidenced by the resonances for a 1,3,4-trisubstituted aromatic fragment at δH8.28 (d,J=1.1 Hz), δH7.14 (d,J= 7.9 Hz), δH7.02 (dd,J= 7.9, 1.1 Hz), and at δC138.0,136.6, 128.5, 128.0, 127.5, 123.4. The connection of the monoterpenyl unit to ring A was confirmed by the HMBC signal from H-10 to C-8 and the long-rangeJ4correlation from H3-18 to C-7. Also, the HMBC correlations (in CD3COCD3) from 5-OH to C-4a, C-5 and C-6 (a methine carbon) further corroborated that the monoterpenyl substitution on ring A was at C-8 instead of C-6, otherwise, 5-OH would have shown HMBC correlations to all quaternary carbons. A comprehensive inspection of the full 2D NMR data (Figure 3) corroborated the constitution structure of 1a/1b as shown, and other important HMBC signals included those from H-2 and H2-3 to C-4, H3-12 to C-10, C-11 and C-13, H3-18 to C-15, C-16 and C-17, H-6′ to C-2. The planar structure of 1a/1b was thus established.

Figure 3. 2D NMR correlations for compounds 1a/1b and 2a/2b.
With the structure of 1a/1b in hand, the structure characterization for 2a/2b then became self-evident. By careful analyses of the NMR data for the two pair of coisolates (Table 1), it was obvious that they incorporated the same flavanone scaffold and only differed from each other at the substitution location of the monoterpenyl unit, which was connected to C-6 rather than C-8 in 2a/2b as supported by the HMBC correlations (in CD3COCD3) from 5-OH to C-4a, C-5 and C-6 (all quaternary carbons, Figure 3). This assignment was also confirmed by the chemical shift difference for 5-OH in 1a/1b (δH12.51) and 2a/2b (δH13.73),which was consistent with the empirical rule that 5-OH in 6-prenylflavonoids is more deshielded than its counterpart in 8-prenylflavonoids [23].
To further investigate the possible transformation of theses interesting molecules, all the samples of 1a/1b, 2a and 2b were further analyzed by the same chiral column after their spectroscopic measurements were completed. As shown in Figure 4, the chiral HPLC chromatograms of them all displayed four peaks like the original mixture, which firmly supported the aforementioned conclusion that the four isomers would undergo a mutual transformation. In order to obtain good NMR data, compounds 1a and 1b (both with limited sample amount) was mixed again after we knew their enantiomeric relationship and measured their other spectroscopic data, so the chiral HPLC analysis here was performed on their mixture. We then proposed a plausible mechanism for this chemical transformation among 1a, 1b, 2a and 2b as shown in Scheme 1. On reviewing the literature, the isomerization at C-2 was once described for sanggenol L and related analogues [23], but the regio-isomerization between C-6 and C-8 substitution had not been reported previously. We speculate that the aromatic monoterpenyl group has extended the conjugation system and could facilitate the formation of the intermediates by stabilizing them.
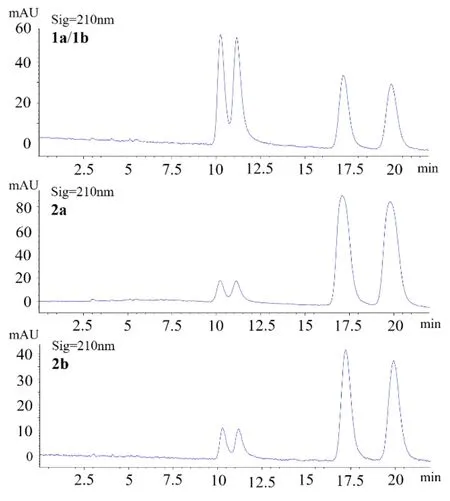
Figure 4. Comparison of chiral HPLC chromatograms for 1a/1b, 2a and 2b.
Hyperglycemia is a major symptom of type 2 diabetes mellitus, and thus controlling hyperglycemia withα-glucosidase inhibitors has been demonstrated to be a strategy for the glycemic modulation and prevention of type 2 diabetes [24].Following our previous work on the exploration of natural α-glucosidase inhibitors from plant resources [18-20, 25] the anti-hyperglycemic effect of 1a/1b,2a and 2b was evaluated by testing thein vitroα-glucosidase inhibitory activity of them, with IC50values of 4.05 ± 0.65, 3.06 ± 0.42 and 6.69 ± 0.51 μM,respectively. These flavanone-monoterpene hybrids showed much better activity than the positive control acarbose which had an IC50of 359.10 ± 2.39 μM.
Supplementary materials
Supplementary Figures S1?S25 are available online atTMR Modern Herbal Medicine. Raw MS, NMR, ECD and [α]Dspectra can be obtained by contacting the corresponding author.
 TMR Modern Herbal Medicine2022年4期
TMR Modern Herbal Medicine2022年4期
- TMR Modern Herbal Medicine的其它文章
- Development status and trend of traditional Chinese medicine database
- Role of traditional herbal medicine in the treatment of malaria
- Clinical observation and experimental study on Kangfuxin fluid in treating indwelling needle-related phlebitis
- Thinking and practice on clinical safety evaluation of combination of traditional Chinese medicine and western medicine
- Phytochemical screening and traditional medicinal potential of Albizia lebbeck (L.) Benth: An update