
Phylotranscriptomic discordance is best explained by incomplete lineage sorting within Allium subgenus Cyathophora and thus hemiplasy accounts for interspecific trait transition
2024-03-09 10:18ZengzhuZhangGangLiuMinjieLi
植物多樣性 2024年1期
Zengzhu Zhang,Gang Liu,Minjie Li
State Key Laboratory of Herbage Improvement and Grassland Agro-Ecosystems,College of Ecology,Lanzhou University,Lanzhou 730000,Gansu,PR China
Keywords: Hemiplasy Multispecies coalescence Lineage sorting Gene tree discordance Phylotranscriptomics Allium subg.Cyathophora
ABSTRACT The transition of traits between genetically related lineages is a fascinating topic that provides clues to understanding the drivers of speciation and diversification.Much can be learned about this process from phylogeny-based trait evolution.However,such inference is often plagued by genome-wide gene-tree discordance (GTD),mostly due to incomplete lineage sorting (ILS) and/or introgressive hybridization,especially when the genes underlying the traits appear discordant.Here,by collecting transcriptomes,whole chloroplast genomes (cpDNA),and population genetic datasets,we used the coalescent model to turn GTD into a source of information for ILS and employed hemiplasy to explain specific cases of apparent “phylogenetic discordance” between different morphological traits and probable species phylogeny in the Allium subg.Cyathophora.Both concatenation and coalescence methods consistently showed the same phylogenetic topology for species tree inference based on single-copy genes(SCGs),as supported by the KS distribution.However,GTD was high across the genomes of subg.Cyathophora:~27%-38.9% of the SCG trees were in conflict with the species tree.Plasmid and nuclear incongruence was also present.Our coalescent simulations indicated that such GTD was mainly a product of ILS.Our hemiplasy risk factor calculations supported that random fixation of ancient polymorphisms in different populations during successive speciation events along the subg.Cyathophora phylogeny may have caused the character transition,as well as the anomalous cpDNA tree.Our study exemplifies how phylogenetic noise can be transformed into evolutionary information for understanding character state transitions along species phylogenies.
1.Introduction
Strong inferences about trait state evolution can be initiated by tracing the number and direction of current trait state evolution from the most likely ancestral trait state(i.e.,homology),if a highly supported species tree among different species is provided.Furthermore,such phylogeny-driven trait evolution can also provide insight into convergence or reversionary evolution (i.e.,homoplasy)(Wake et al.,2011;Guerreroa and Hahna,2018).However,phylogeny-based trait inference is often limited by the widespread occurrence of gene-tree discordance (GTD).GTD has been widely found in organismal phylogenies,especially in phylogenomics,where thousands to millions of genes are typically used to reconstruct phylogenies.The rampant GTD,especially when the incongruent traits occur in these discordant gene regions,known as hemiplasy(Guerrero and Hahn,2018;Wu et al.,2018),can amplify false trait state inference even in highly supported species trees.However,although such effects may be intrinsic and profound(Wu et al.,2018),few studies have documented the effects of gene tree discordance on phylogeny-based trait state inference.
The most common explanations for GTD are incomplete lineage sorting (ILS) and introgressive hybridization (IH) (Degnan and Rosenberg,2009),aside from gene tree estimation error which likely was caused by a paucity of informative sites at selected gene loci,misidentification of orthologs due to paralogy,and poor fit of the substitution model to the sequence data.ILS is strongly influenced by effective population size and evolutionary time (Pamilo and Nei,1988),as it occurs through the random transmission of segregating ancestral variation between successive nodes in a species tree (Maddison,1997).As a consequence,ILS would arguably be exaggerated in groups that underwent recent fast radiations(e.g.,Pease et al.,2016; Wang et al.,2018; Wu et al.,2018; Li et al.,2021).However,IH can also lead to GTD when ancestral or recent loci are shared between diverging lineages and there is a conflict between evolutionary history of introduced genes and those from native genomes (e.g.,Wu et al.,2018; Feng et al.,2019).If ILS is a major source of GTD across the phylogeny,the probability of hemiplasy for incongruent trait evolution along a targeted phylogeny should be considered (Robinson and Anne,2011; Mendes et al.,2016; Hahn and Nakhleh,2016; Copetti et al.,2017;Guerrero and Hahn,2018; Wu et al.,2018),rather than simply mapping the states of traits on a single species tree to deduce their evolutionary trajectory (Wake et al.,2011; Guerrero and Hahn,2018).In particular,it is important to note that if the genes underlying trait variation occur in these discordant genomic regions,unrelated species are likely to coalesce,potentially leading to incorrect inferences about ancestral trait states.When successive speciation events and associated lineage sorting of genetic polymorphisms leads to a discordance between species and gene trees,this is known as hemiplasy (Avise and Robinson,2008).In this sense,hemiplasy is a genuine form of trait homology (due to common ancestry)in a species tree,but it is still not homoplasy at the level of the gene tree (Avise and Robinson,2008; Wu et al.,2018).To better explain the evolution of incongruent traits when GTD occurs,Guerrero and Hahn (2018) developed a method,the hemiplasy risk factor(HRF),to highlight individual branches of the phylogenetic tree along which hemiplasy is likely to occur and contrast them with the probability of convergence of the observed incongruent trait.Thus,quantifying the HRFs in a coalescent unit tree can help us explore the evolution of many incongruent states of traits of interest.
The phylogenetic relationship between the monotypic Himalayan genusMilulaPrain and the globally distributed genusAlliumL.has been widely debated.The taxonomic notation ofMilulahas always been problematic due to the conspicuously elongated spicate compared to the mostly umbel or capitate inflorescences inAllium(Krause,1930;Hutchinson,1959; Stearn,1960;Traub,1972;Takhtajan,1987; Friesen et al.,2000).The growing body of integrated evidence strongly supports the revision of the monotypic speciesM.spicataPrain toA.spicatum(Prain) N.Friesen (Friesen et al.,2000,2006).Molecular evidence suggests thatA.spicatumbelongs to subg.CyathophoraR.M.Fritsch,which consists of five diploid species,includingA.maireiLˊeveillˊe,A.rhynchogynumDiels,A.spicatum,A.cyathophorumBureau&Franch.,andA.farreriStearn,and one tetraploid species of hybrid origin(A.tetraploideumM.J.Li& X.J.He) (Huang et al.,2014; Li et al.,2019a).Despite the abundance of evidence supporting the genetic affinities betweenA.spicatumand its relatives,the occurrence of GTD confounds evolutionary trajectories of traits between them.For example,A.maireiandA.spicatumare successively sister to the most recently diverged sister speciesA.cyathophorumandA.farreriin the nuclear ITS tree; however,A.spicatumandA.farrericonstitute the most recently diverged sister species,andA.maireiandA.cyathophorumare successively sister to them in the chloroplast DNA(cpDNA)tree and the low copy nuclear gene(At103)tree(Li et al.,2016).In many studies,such GTDs have been treated as phylogenetic noise(Knowles and Kubatko,2010),and only a few studies have explained the underlying mechanisms of such GTDs and evaluated the impact of such GTDs on the phylogeny-based inference of trait states.
Here,using 1262 single-copy genes(SCGs),chloroplast genomes and population genetic data,we further investigated GTD across the phylogeny ofAlliumsubg.Cyathophora.Furthermore,we used coalescent simulations to explore the underlying mechanisms of such GTD within subg.Cyathophoraand turned the clearly present GTD into an information source of ILS to elucidate the controversial character evolutionary history of subg.Cyathophora.
2.Materials and methods
In order to clearly illustrate the operating procedures in this study,a simple analytical flowchart has been generated in Fig.S1.The detailed information is described as follows.
2.1.Transcriptome and chloroplast genome collection
In this study,a total of five species were selected,including four diploid species (Allium mairei,A.spicatum,A.cyathophorum,andA.farreri)ofAlliumsubg.Cyathophora,and one diploid individual ofA.przewalskianumRegel,which was used as an outgroup.Sampling information is provided in Table S1.The transcriptomes and chloroplast genomes of these five species were obtained from NCBI under accession numbers SAMN13698330-SAMN13698335 and MN882559-MN882564,respectively(all from Li et al.,2021).
2.2.Chloroplast assembly
Fast-Plast v.1.2.6 (https://doi.org/10.5281/zenodo.973887) was used to assemble the whole chloroplast genomes (cpDNA).Geneious v.10.2.6 (Kearse et al.,2012) was then used for manual validation and consensus sequence generation.The assembled chloroplast genomes were aligned using MAFFT (Katoh and Standley,2013).
2.3.Transcriptome assembly and SCGs identification
The paired-end clean reads of each transcriptome werede novoassembled using Trinity v.2.1.5 (Grabherr et al.,2011) with default parameters.TRANSDECODER-v.5.0.2 was used to predict the longest putative CDS and protein sequences in the open read frame(ORF).CD-HIT (Huang et al.,2010) was used to generate the nonredundant CDS and protein sequences with a threshold of 0.95.OrthoMCL (Li et al.,2003) was used to identify orthologous sequence groups for the five species,and then SCGs were extracted for downstream analyses using a custom Perl script.Each orthologous sequence group was aligned using PRANK (L¨oytynoja and Goldman,2008) with the ‘-codon’ parameter.The orthologous clusters were filtered by removing those with length <300 bp and‘-’ character in each sequence >50%.
2.4.Phylogenetic analysis
The concatenation and coalescence methods were used to infer the species tree.For the concatenation approach,all SCGs were concatenated into a supermatrix with the alignment length in 950,648 bp,and then a maximum likelihood(ML)tree was inferred in RAxML v.8(Stamatakis,2014)using the GTR-GAMMA model and‘-f a’for rapid 100 bootstrapping to search for the ML tree with the highest score(hereafter referred to as the nuclear ML tree).For the coalescent approach,we first reconstructed individual ML trees for each of the SCGs in RAxML with the same parameters as described above.After filtering the SCG trees with bootstrap support(BS)less than 70% at each node using a custom Perl script,we used the remaining SCG trees to infer a coalesced tree in MP-EST 2.0 (Liu et al.,2010a) with a maximized pseudo-likelihood function(henceforth referred to as the nuclear MP-EST tree).Similarly,a ML tree based on the aligned chloroplast genomes was also inferred in RAxML v.8(hereafter called cpDNA tree).We then inputted the SCG trees with BS ≥70% and the topology of the cpDNA tree into MPEST 2.0 to generate a cpDNA MP-EST tree with the same branch length in coalescent units and topology as the cpDNA tree.The“tree.noclock2clock” function in the R package phybase (Liu et al.,2010b) was used to convert a non-clocklike tree into a clocklike(ultra-metric)tree using an ad hoc approach.Densitree(Bouckaert,2010) was used to describe all ultra-metric trees together to illustrate phylogenetic heterogeneity.
2.5.Synonymous substitutions per synonymous site (KS)
To further validate the phylogenetic relationships among the target species,we calculated the percentage of synonymous substitutions per synonymous site (KS) between given pairwise orthologs (e.g.,Guan et al.,2019).We used INPARANOID v.4.1.1(Sonnhammer and ¨Ostlund,2015)to infer interspecific orthologous sequences,and single-copy orthologs were extracted using a custom Perl script.All pairwise orthologous sequences were aligned using PRANK with the ‘-codon’ parameter.Then,a ML method was implemented in the YN00 program of the PAML package v.4.1 (Yang,2007) to compute KSbetween orthologous sequence pairs.The resulting KSwere plotted as a histogram (KSplot) with KS≥1.0 removed due to sparse data.We implemented Gaussian mixture distributions using an expectation-maximization(EM) algorithm to infer significant peaks in each observed KSdistribution in the R package mixtools (Benaglia et al.,2009).A parametric bootstrap(B=100)of the likelihood ratio statistic was performed using the “boot.comp” function in the R package mixtools to examine the most likely number of Gaussian components that fit each KSdistribution.Sizer tests (Chaudhuri and Marron,1999) were used to further confirm the Gaussian mixture model components across a distribution at various (log transformed)bandwidths to distinguish true data features from noise by visually testing between increases,decreases,or values not significantly different from zero.True peaks were identified as those Gaussian plots that shifted significantly from increase to decrease in the sizer.The significant (alpha = 0.05) features in each observed KSdistribution were tested when KSvalues were ≤1.0 and KSbandwidths were 0.01-1.00.The corresponding age of each KSplot was calculated using the formula: KS/generation time/mutation rate,with a generation time of two years and an average mutation rate of 1.25 × 10-8(which varies from 1 × 10-8to 1.5 × 10-8) for plants(Ossowski et al.,2010).
2.6.Population phylogenies based on ten nuclear SCGs
To further confirm the widespread occurrence of GTD withinAlliumsubg.Cyathophora,ten SCGs(9266-2,9201-5,10076-6,9341-10,10096-12,9360-14,10110-18,9259-23,7189-26,and 7214-27)were downloaded from NCBI under accession numbers MT479228-MT481754 (all from Li et al.,2021) for population phylogenetic analyses.A total of 46 individuals were sampled,including 18A.cyathophorum,22A.farreri,five A.spicatum,and oneA.mairei(Table S1).Alignment of each gene was performed in MEGA 6.0 (https://www.megasoftware.net/) and checked by eye.The haplotype file for each gene was generated in DNAsp v.6.0(http://www.ub.edu/dnasp).The ML tree for each haplotype file was reconstructed in PhyML (Guindon,2010) using a likelihoodbased method.
2.7.Coalescence simulations
To test whether ILS was a major factor causing GTD across theAlliumsubg.Cyathophoraphylogeny,we performed coalescent simulations following the method developed by Wang et al.(2018;https://github.com/wk8910/ILS_simulation).In brief,we started by counting the topology of the SCG trees using the R package ape(Paradis and Schliep,2019).Then,we simulated 500,000 gene trees for both the nuclear and cpDNA MP-EST trees under the multispecies coalescent model using the “sim.coaltree.sp” function in R package phybase.Several approaches were used to test the topological consistency or difference between the observed SCG trees and the simulated gene trees.First,we compared the distributions of the topological frequencies of the observed SCG trees and the simulated gene trees.Second,we fitted a linear regression equation in R for the frequencies of the observed SCG trees and the probabilities of the simulated gene trees.Third,we first calculated the Robinson-Foulds (RF) distances from each of the observed SCG trees respectively to the nuclear (RF1) and cpDNA (RF2) MP-EST trees using the “tree.distance” function in the R package phybase,and then calculated ΔRF=RF1-RF2 to measure whether each SCG topology was more similar to the nuclear MP-EST tree or the cpDNA MP-EST tree.Finally,a likelihood ratio test(LRT)was performed to assess the goodness of fit of the two simulated gene tree distributions to the observed gene tree distribution by testing the statistic t = -2(L(τn) - L(τm)) with 1000 parametric bootstrap replicates,where τm was a null hypothesis about the goodness of fit of the simulated cpDNA trees to the observed SCG trees,τn was an alternative hypothesis about the goodness of fit of the simulated nuclear gene trees to the observed SCG trees,and L(τm) and L(τn)were their respective log-likelihood tests.
For our coalescent simulations,we expected a higher frequency of the topology same with the cpDNA MP-EST tree if ILS alone could explain the topological incongruence between the cpDNA and nuclear trees.In contrast,an absence or lower frequency of the topology same from the cpDNA MP-EST tree would be expected if IH was present in the evolutionary history of subg.Cyathophora.We therefore counted the number of additional lineages for both the observed SCG trees and the simulated nuclear gene trees using the“deep_coal_count”function in phyloNet v.2.4(Than et al.,2008).If IH had occurred in the evolutionary history of subg.Cyathophora,we would expect to see more extra lineages in the observed SCG trees than in the simulated nuclear gene trees.To further test whether IH contributed to GTD within subg.Cyathophora,we estimated interspecific relationships by analyzing all observed SCG trees using parsimonious inference of hybridization in the presence of ILS (Yu et al.,2013) in phyloNet,assuming one versus two past hybridization events.
2.8.Reconstructing ancestral trait state
Phylogeographic investigation showed that spicate and umbel inflorescences are clearly geographically segregated across the 500 mm Qinghai-Tibet Plateau isohyet (QTP,Li et al.,2019b).Accordingly,their ecological niche (here represented by altitude),seed size,flowering phase,and ovary size also differ(Li et al.,2016).Among these traits,geographic distribution (western QTP/eastern QTP) and inflorescence (spicate/umbel) are categorical; seed size(thousand seed weight:1.317,0.948,0.878,1.691,2.009 g),flowering phase(month:6-9,8-10,8-10,6-8,6-8),ovary size(3.3,2.9,2.1,4.0,4.2 mm)and altitude(mean values:3400,2287,4219,3899,2703 m) forA.przewalskianum,A.mairei,A.spicatum,A.cyathophorumandA.farreri,respectively,are quantitative traits(Table S2).These traits are considered irreversible because they are stable in the natural populations.We then used the Bayesian Binary Method (BBM) in RASP (Yu et al.,2020) based on the nuclear ML tree to infer the ancestral state of the categorical traits.We ran a total of one million generations,nine hot Markov chains and one cold chain with temperature increments of 0.1 for each analysis.For quantitative traits,we first calculated their square root values and then used the“fastAnc”function in the R package phytools(Revell,2012) to quickly estimate their ancient state based on the nuclear ML tree.
2.9.Identifying genes under natural selection
To examine the effect of natural selection on spicate-specific lineages throughout the phylogeny ofAlliumsubg.Cyathophora,we calculated ω (dN/dS ratios),a criterion of selection pressure,using the CODEML program in the PAML package on the observed SCG trees with BS ≥70%.In general,ω = 1 indicates neutral selection,ω >1 positive selection,and ω <1 negative selection(Yang,2007).The ω values were calculated by implementing a null model(Ma0,no lineages with specifically high nonsynonymous substitutions due to natural selection) and an alternative model (Ma,spicate was counted as a lineage-specific trait and was likely under positive selection,which consequently could have led to GTD) on each of the SCG gene trees using a strict branch-site approach used to estimate and predict recent radical evolution(Zhang,2000).The LRTs were used to estimate the significance of ω values(P<0.05)by calculating the chi-squared(χ2)distribution between 2△L and df.The significancePvalue was adjusted using the false discovery rate(FDR).The positively selected sites were identified using Bayes Observed Bayes (BEB) with posterior probability less than 0.08(Yang et al.,2005).
2.10.Quantifying and validating hemiplasy
ILS and neutral selection are two important assumptions for quantifying hemiplasy(Guerreroa and Hahna,2018).Therefore,we just used the SCGs under neutral selection to infer a MP-EST tree(hereafter called neutral MP-EST tree).In the present of ILS,the Hemiplasy Risk Factors (HRFs) were calculated and visualized for the internal nodes of the neutral MP-EST tree using R package pepo(https://github.com/guerreror/pepo),where an average mutation rate of 1.25 × 10-8(the substitution rate per nucleotide site per year,s/s/y) for plants (ranging from 1 × 10-8to 1.5 × 10-8;Ossowski et al.,2010),and the effect population size(Ne)of 133,870(with 95%highest posterior density interval 6310-978,952)for the most recently common ancestor of subg.Cyathophora(Wang et al.,2015)were used to calculate the 2Nemutation rate of 3.21×10-3,which varied from 1.5 × 10-4to 2.35× 10-2.
To evaluate the role of hemiplasy on the incongruent characters along the phylogeny ofAlliumsubg.Cyathophora,we selected the genes likely to be correlated with inflorescence development to infer tree topologies.We first ran the SCG functions with the default parameters using the TrEMBLEMBL database(http://kr.expasy.org/sprot/) as a background.To further detect the biological processes of the SCGs,Gene Ontology (GO) enrichment was performed.R package clusterProfiler (Yu et al.,2012) was used for GO pathway enrichment analysis.The GO background of the genome of the species was obtained using the eggNOG mapper (Huerta-Cepas et al.,2017) with the default parameters.To further find genes related to inflorescence development,we used Interproscan(https://www.ebi.ac.uk/interpro/about/interproscan/) to search all SCGs to map genes of the ARF (auxin response factor) family,the SST family (Li,2012),the PLC (phosphoinositide phospholipase C)family (Li et al.2018),the FT/TFL1 (flowering locus T/terminal flower 1) family,the SPB (squamosa promoter binding protein)family,the MADS box,and the PEBP (phosphatidy lethanolaminebinding protein)family,respectively.
3.Results
3.1.Transcriptome assembly
Clean sequences comprising of 7.34,7.67,6.76,6.67 and 13.1 Gb cDNA bases were obtained forAllium cyathophorum,A.farreri,A.spicatum,A.maireiandA.przewalskianum.Thede novoassembly produced 469,514,368,926,267,859,300,360 and 477,740 contigs with N50 values of 833,1157,736,957 and 1178 bp for these species,respectively(Table S3).
3.2.Phylogeny of Allium subg. Cyathophora
A total of 1262 SCGs were identified using OrthoMCL for the four diploid species ofAlliumsubg.Cyathophora,withA.przewalskianumused as the outgroup.Among these 1262 SCG trees,840 were found to have BS ≥70%.The consistent topology between the concatenation and coalescence-based trees (Fig.S3) strongly supported thatA.cyathophorumandA.farreriwere the closest species,andA.maireiandA.spicatumwere successively sister to them (Fig.1B and C).However,the cpDNA tree revealed thatA.spicatumandA.farreriwere the closest relatives,and they were each sister toA.cyathophorum,and finally the three were sister toA.mairei(Fig.1D).This was consistent with the topology based on the cpDNA fragments (Li et al.,2019a,b).Ultimately,the nuclear and cpDNA trees were clearly phylogenetically incongruent.

Fig.1.Phylogenetic relationships among the four diploid species of the genus Allium subg.Cyathophora based on transcriptome analyses.(A) The density plots of synonymous nucleotide substitutions (KS) of the pairwise orthologs.The divergence time was estimated using the formula: KS/generation time/mutation rate.(B) The ML tree based on the concatenation of 1262 SCGs.(C)The MP-EST tree based on 840 SCG trees with the bootstrap support(BS)≥70%at each node.(D)The ML tree of the whole-chloroplast genome.BS at each node in these three trees are 100%.
3.3.KS-based species divergence
The species pairwise orthologs identified by INPARANOID were 13,525,13,097 and 12,761 inAllium mairei-A.spicatum,A.mairei-A.cyathophorumandA.mairei-A.farreri,respectively;the pairwise orthologs inA.spicatum-A.cyathophorumandA.spicatum-A.farreriwere 14,109 and 13,711,respectively;and the pairwise orthologs ofA.cyathophorum-A.farreriwere 15,204 (Table S4).Our Gaussian mixture modeling identified a unique peak for synonymous site divergence (KS) for each pairwise species,and each was significantly supported by the Sizer tests (Fig.S2 and Table S5).The interspecific divergence ages for inA.mairei-A.spicatum,A.mairei-A.cyathophorumandA.mairei-A.farreriwere respectively estimated to be approximately 4.80(4.00-6.00),4.24(3.53-5.30)and 4.48 (3.73-5.60) million years ago(Ma),respectively based on the corresponding KSpeak values of 0.120,0.106,and 0.112; the divergence ages forA.spicatum-A.cyathophorumandA.spicatum-A.farreriwere at ~3.32 (2.77-4.15) and ~3.44 (2.87-4.30) Ma respectively,with the corresponding KSvalues of 0.083 and 0.086;the divergence time ofA.cyathophorumandA.farreriaround at 1.68(1.40-2.10) Ma,with the KSpeak of 0.042 (Fig.1A and Tables S5-S6).Such KS-based interspecific divergence,such that the closer the phylogenetic relationships,the smaller the KSvalues corresponding to the latter divergence times,also provided support for the phylogeny of subg.Cyathophorarevealed by the SCGs(Fig.1).
3.4.Genome-wide GTD
Among the SCG trees,a high degree of phylogenetic discordance was found:60.90%of all SCG trees were congruent with the nuclear MP-EST tree,while the remaining 30.91% of trees showed discordant topologies with the nuclear MP-EST tree(Fig.2A and Table 1).A similar result was also observed among the SCG trees with BS ≥70% (Table 1).All SCG tree topologies were classified into 15 topological types (Table 1).According to the phylogenetic position of the spicate-specificAllium spicatum,these 15 types were further divided into four classes:Class I,1 out of 15 types,agreed with the nuclear MP-EST tree in whichA.spicatumwas directly sister to theA.cyathophorum-A.farrericlade;Class II,6 out of 15 types,showed thatA.spicatumwas sister to eitherA.cyathophorumor toA.farreri;Class III,2 out of 15 types,showed thatA.spicatumwas the most distant species in the phylogenies,orA.spicatumwas clustered together withA.mairei,respectively;and Class IV,6 out of 15 types,exhibited thatA.maireiwas sister toA.farreri,A.cyathophorumorA.spicatum,respectively,at the most recent node in the phylogenies (Table 1).The SCG trees with the second highest proportion(12.12%of all SCG trees;9.76%of the SCG trees with BS ≥70%)had the same topology as the cpDNA tree (A.spicatumandA.farreriwere the closest relatives at the most recent branch).Notably,the proportion of the gene trees resembling the cpDNA tree was slightly higher than the proportion of gene trees in whichA.spicatumwas directly sister toA.cyathophorumat the most recent node (11.25% of all SCG trees; 7.38% of the SCG trees with BS ≥70%)(Table 1).Furthermore,A.maireiwas always positioned in the earliest diverged branch in the top three SCG tree topologies,accounting for the highest proportion(61.09%+12.12%+11.25%of all SCG trees; 72.98% + 9.76% + 7.38% of the SCG trees with BS ≥70%)(Table 1).

Table 1 Four classes of the observed tree topologies with their frequencies,observed frequencies among trees with higher bootstrap support(BP ≥70%),and simulated frequencies.The Class I indicates gene-tree topology consistent with the nuclear species tree,with no ILS affection;the other three classes indicate ILS links the spicate Allium spicatum to different umbel lineages,respectively.Pr,A.przewalskianum; Ma,A.mairei; Sp,A.spicatum; Cy,A.cyathophorum; Fa,A.farreri.
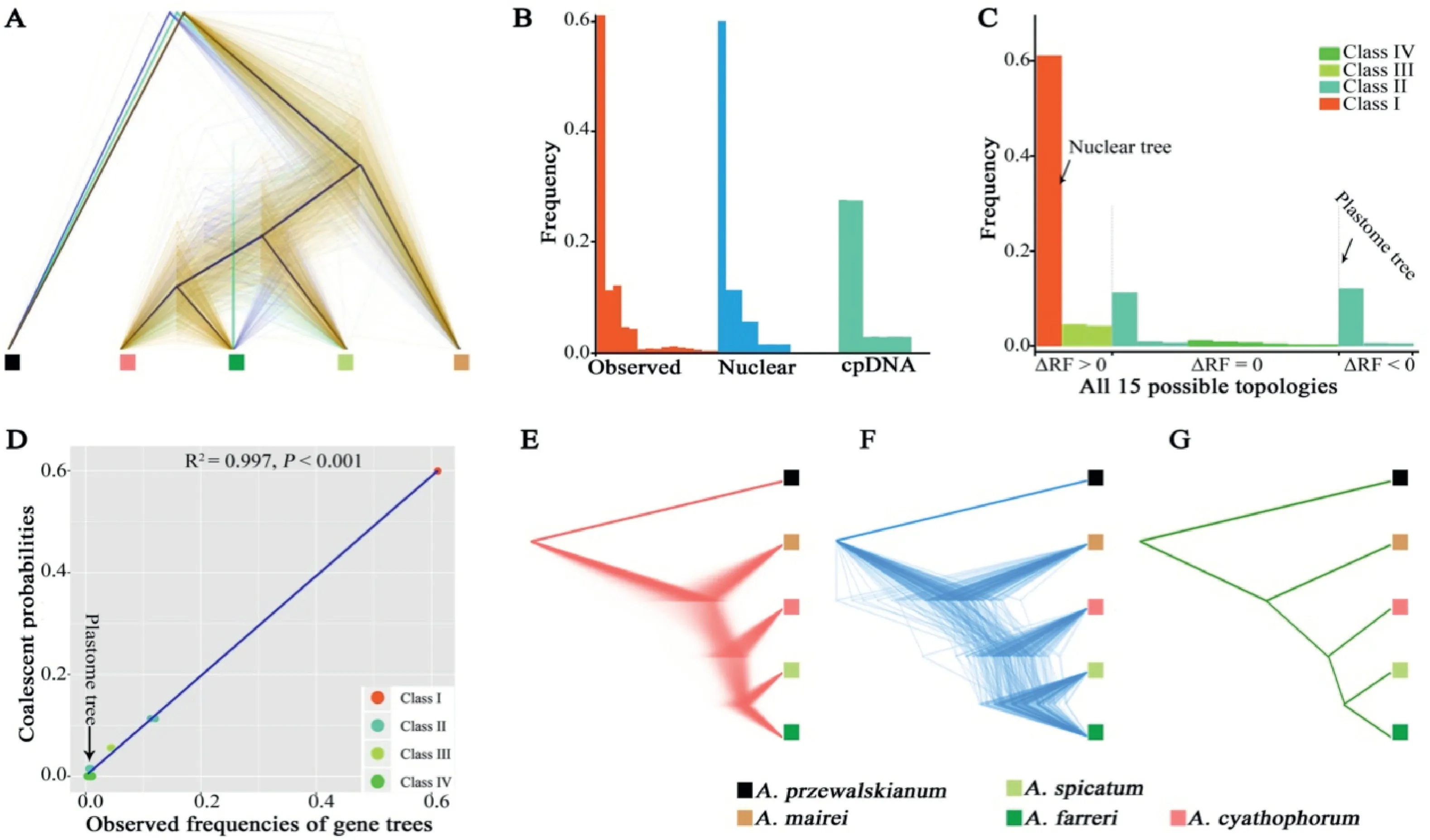
Fig.2.Incomplete lineage sorting (ILS)model for gene-tree discordance (GTD)across the phylogeny of Allium subg.Cyathophora,using A.przewalskianum as an outgroup.(A)The densitree of 1262 SCG trees.Black lines indicate the consensus tree.Yellow lines indicate the most frequent topology,followed by the second and the third ones indicted by blue and green lines (Table 1).(B) The distributions of the observed 1262 ML SCG trees,the simulated gene trees respectively from the nuclear and cpDNA MP-EST trees,respectively.(C)Ordering of the empirical frequencies of all 15 possible gene tree topologies according to the frequency of each topology in ΔRF >0(gene trees more similar to the nuclear MP-EST tree),ΔRF=0(trees equally similar to the nuclear and cpDNA MP-EST trees)and ΔRF <0(trees more similar to the cpDNA MP-EST tree).All 15 possible topologies are classified into four classes(detail in Table 1)and are indicated by different colors.(D)A fitted linear regression between the simulated frequencies of gene topologies of different classes based on the coalescence model and the corresponding observed frequencies.The arrow indicates the cpDNA topology.(E)Simulated gene trees with the same topology as the cpDNA tree.(F)Observed gene trees having the same topology as the cpDNA tree.(G)Observed cpDNA tree using the whole chloroplast sequence.All trees in(E,F,G)are scaled to the same total length.
3.5.Population phylogenies based on ten SCGs
To further validate the phylogenetic discordance at the population level,we obtained sequence variations at ten SCG loci to reconstruct the phylogenies of multiple individuals for each ofAllium spicatum,A.cyathophorumandA.farreri,all of which have high phylogenetic discordance,and an individual ofA.maireiwas used as an outgroup.The resulting phylogenies were highly consistent with the phylotranscriptomic analyses.No common haplotypes were found among these species (Figs.2A and 3).We found that six (10096-12,7189-26,9360-14,7214-27,9266-2 and 9201-5)of the ten gene trees were topologically consistent with the nuclear MP-EST tree,in whichA.maireiandA.spicatumwere successively sister to the most recently diverged speciesA.cyathophorumandA.farreri(Fig.3).Among the remaining four gene trees,two(9341-10 and 10076-6)showed thatA.spicatumandA.cyathophorumwere joined together at the youngest node,and both were sister toA.farreri.In the other two trees (9259-23 and 10110-18)A.spicatumandA.farreriwere clustered together in the youngest node,and then both were sister toA.cyathophorum,and finally the three toA.mairei(Fig.3).
3.6.Multispecies coalescent simulations of ILS for the GTD
Our statistical test showed that the distribution of the simulated gene trees based on the nuclear MP-EST tree had a much better fit to the distribution of the observed SCG trees than the simulated gene trees generated from the cpDNA tree (Fig.S4).In short,a striking agreement between the distributions of simulated nuclear trees and observed SCG trees was found when simulating 500,000 gene trees each based on the nuclear MP-EST tree and the cpDNA tree just under ILS alone(Fig.2B and Table 1).We found that ΔRF >0 occurred in the majority of the observed SCG trees (~70.0%),suggesting that they had the same topology as the nuclear MP-EST tree.However,the proportion of SCG tree topologies closer to the cpDNA tree with ΔRF <0 was small,about 13.3%,and the remaining 16.7%of all gene trees had the same RF distance(ΔRF=0)from the nuclear MP-EST tree and the cpDNA tree(Fig.2D).
The overall correlation coefficient for the coalescently simulated nuclear trees and the observed SCG trees was 0.997 (P<0.001,Fig.2D),which was very close to 1,indicating a good fit between the multispecies coalescent simulations and the observed SCG trees.The distributions of pairwise tree distances between the nuclear MP-EST tree and the gene tree for both the observed and simulated nuclear trees showed a clearly consistent pattern (Fig.S4).Furthermore,both the observed (30.71%,258/840) and simulated(30.11%,150,553/500,000)gene trees based on the nuclear MP-EST tree showed approximately the same number of extra lineages.All these results consistently suggested that ILS was a major factor of genome-wide GTD within subg.Cyathophora.Interestingly,among the simulated gene trees based on the cpDNA tree,we found that the proportion of gene trees like the cpDNA tree (27.52%) was approximately equal to that like the nuclear MP-EST tree (27.60%;Fig.2B and Table 1).Our converted gene tree branches of the cpDNA-like tree were very similar to those of the cpDNA tree in both the observed and simulated gene trees (Fig.2E,F,G).Both results supported that the anomalous cpDNA tree was a product of ILS.
We performed phylogenetic network analysis to further exclude the causal role of IH in genome-wide GTD.Our phyloNet-based phylogeny was consistent with the nuclear MP-EST tree if a past hybridization event occurred in the evolutionary history of subg.Cyathophora,in which no hybridization was found between the spicate-specificAllium spicatumand the other umbel species.However,an ancient genomic exchange from the common ancestor of the clade ofA.spicatum-A.cyathophorum-A.farreritoA.maireiwas detected with a small fraction of 17.27%(Table S7).When two past hybridization events occurred,a spurious phylogenetic network was generated that was highly discordant with the nuclear MP-EST tree,and thus we rejected the two hybridization event hypothesis (Table S7).
In conclusion,our multispecies coalescent simulations and phyloNet-based network analyses both support that ILS is the major cause of genome-wide GTD,as well as the phylogenetic incongruence between cpDNA and nuclear.
3.7.Genes under the positive selection
Depending upon the branch with spicate being tested across 840 SCG trees with BS ≥70%,some selection was identified on~22.5%of all loci(189 out of 840);genes where ω >1 andP<0.05 were ~5.24% of all loci (44 out of 840),and genes with ω >1 andP<0.01 were ~2.98%(25 out of 840)in our dataset(Table S8).Five tree topological types were observed in these positively selected gene sites and they coincided with three of the four classes(Table S9).Interestingly,the proportion of each class in these 44 positively selected sites was highly consistent with their proportion in 840 SCG trees: class I accounted for 72.7%,class II for 18.2%(11.4% + 6.8%) and class III for 9.1% (6.8% + 2.3%) (Table S9); their corresponding abundances in 840 SCG trees were 72.98%,18.34%(9.76%+7.38%+0.36%+0.48%+0.36%)and 7.14%(3.57%+3.57%),respectively (Table 1).With respect to these results,we found no significant difference between the proportions of genes under positive selection within the nuclear MP-EST like trees (5.22%,32/613) and within the cpDNA-like gene trees(5.19%,8/154).
Gene Ontology(GO)enrichment showed that the gene functions of these positively selected genes were enriched in different molecular biological processes(Table S9).The positively selected genes that showed discordant topologies with the nuclear MP-EST tree,were enriched in detoxification proteins,oleosin,transmembrane proteins,chromosome metabolism,cell cycle control,binding and catalytic activity (Table S9).
3.8.Ancestral trait states and hemiplasy for the incongruent traits
The RASP-based ancestral trait inferences strongly indicated(BS=100%)that the ancestral inflorescence in each internode was most likely in the umbel state and distributed in the eastern QTP(Fig.4A).In short,we found that the most recent common ancestor(MRCA) ofAllium cyathophorumandA.farreriwas 99.53%umbel + 0.47% unknown; the MRCA ofA.spicatumandA.cyathophorum-A.farreriwas 86.98%umbel+13.02%spicate;and the MRCA of the subg.Cyathophorawas 96.13% umbel +3.87% unknown.For the ancestral states of the quantitative traits,different values were quantified for each of the branch tips: 1)A.spicatumhas the smallest seed size,which is similar toA.mairei,but significantly different with the larger seed size inA.cyathophorum,and the largest one inA.farreri; 2)A.spicatumshows a later flowering phase from August to October,which is similar toA.mairei,but different with the earlier flowering phase ofA.cyathophorumandA.farreri,whose flowering period ranges from June to August;3)A.spicatumis distributed in the regions with the highest altitudes,which is similar toA.cyathophorum,but different fromA.farreriandA.mairei,which are chiefly distributed across lower altitudes;4)A.spicatumhas the smallest ovary size,which is different with the intermediate ovary size inA.mairei,and the largest ovary size inA.cyathophorumandA.farreri.We observed that trait values of the branch tips mostly were near end of observed value ranges,whereas the trait values of the internal nodes were nearer the mean of trait distributions (Fig.4B).These results suggest that an ancestral trait may have evolved along two contrast directions to form derived traits.

Fig.4.Trait evolution and estimation of HRFs for hemiplasy.(A-B) The phylogeny-based ancestral trait inferences for the (A) categorical traits (inflorescence and geographical distribution) and (B) quantitative traits (seed size,flowering phase,altitude,and ovary size).Numbers on the branches indicate the ancestral trait values.(C-E) Quantification of hemiplasy for trait transition along the phylogeny with constant substitution rate (1.25 × 10-8) and different 2Ne (C) 1.5 × 10-4 (D) 3.21 × 10-3 and (E) 2.35 × 10-2.(F) A representative tree topology of the ARF gene family and (G) one of the PLC gene family,probably related with inflorescence development.
We calculated the HRFs for each internal branch of the neutral MP-EST tree in the presence of ILS.For a given substitution rate per nucleotide site per year(s/s/y)of 1.25×10-8,we found HRF values decreased with increasing effective population size.In short,when parameterized with the lowest 2Nemutation rate,we observed the highest values of HRF of 0.988 at the internal branch ofAllium cyathophorumandA.farreri,and 0.960 at the internal branch ofA.spicatumandA.cyathophorum-A.farreri(Fig.4C).Similarly,when using the median 2Nemutation rate,HRF at the internal branch ofA.cyathophorumandA.farreriwas 0.793,and the HRF for the internal branch ofA.spicatumandA.cyathophorum-A.farreriwas 0.525 (Fig.4D).Perhaps unsurprisingly,parametrizing with the largest 2Nemutation rate returned the lowest HRFs:0.331 at the internal branch ofA.cyathophorumandA.farreri,and 0.121 at the internal branch ofA.spicatumandA.cyathophorum-A.farreri(Fig.4E).
Gene Ontology (GO) enriched the neutral genes in biological process,cellular component and molecular function (Fig.S5).The ARF (auxin response factor,they bind to auxin response DNA elements in the promoters of auxin-regulated genes and either activate or repress transcription of these genes depending on a specific domain in the middle of the protein) and PLC (phospholipase C)families have been found to play key roles in regulating plant growth and development.We found that two genes each from the ARF and PLC families,respectively,showed the topologies discordant with the species tree (Fig.4F and G).In these two trees,A.spicatumwas either linked toA.farreri(Fig.4F) or toA.cyathophorum(Fig.4G).
4.Discussion
4.1.ILS for the genome-wide GTD
Phylotranscriptomics has been widely used for phylogenetic analyses by extracting reliable orthologs from assembled transcripts (Cheon et al.,2020).This approach greatly improves our ability to infer species phylogenies when considering species with giant genomes such as theAlliumspecies (Sun et al.,2020).Our phylotranscriptomics of the four diploid species of the genusAlliumsubg.Cyathophorayielded a strongly supported species relationship using both concatenation and coalescence methods.Thus,A.cyathophorumandA.farreriare the most closely related species,both are sister to the spicate-specificA.spicatum,and the three are sister toA.mairei(Fig.1B and C).Our KS-based species divergence also demonstrated the reliability of this species phylogeny,indicating that the earliest diverged species wasA.maireiaround 4.8 Ma,the second species to diverge wasA.spicatumaround 3.44 Ma,and the most recently diverged species areA.cyathophorumandA.farerriaround 1.68 Ma(Fig.1A).These divergence times are similar to those inferred from the constant mutation rate(Li et al.,2019b).However,widespread gene tree disagreement(GTD,~38.9%of 1262 SCG trees)was detected among orthologous gene trees,even those with high BS across all nodes(~27%of 840 SCG trees with BS ≥70,Fig.2A).The GTD observed in the phylotranscriptomics was also detected in the population phylogeny (4 out of 10 SCGs,Fig.3),similar to the discrepancies in the cpDNA and nuclear gene trees(Fig.1; Li et al.,2016).
Our previous (Li et al.,2019b) and current (Fig.3) population genetics both indicated that no shared haplotypes were found among theAlliumsubg.Cyathophoraspecies,suggesting little gene flow among them.Our phyloNet-based phylogenetic network analysis also supported that (Table S7).We identified similar proportions of gene tree topology classes in positively selected and empirical genes,suggesting natural selection has little influence on the GTD associated with the subg.Cyathophoragenomes (Table 1 and S9).The distribution of the observed gene trees showed that GTD occurred mainly among the recently divergedA.spicatum,A.cyathophorumandA.farreriprobably due to ILS (Class II,accounting for 82% of all the discordant gene trees),although individual gene trees presented highly variable bootstrap support for the specific position of individual species (Table 1).Indeed,our coalescent stimulations also strongly indicated that ILS could have linkedA.spicatumtogether with different species (Fig.2 and Table 1).The origin time of subg.Cyathophoracoincided with the dramatic uplift of the QTP and associated ancient climate in the region (the eastern QTP; Fig.4A; Li et al.,2019b).These geological events could have led a population bottleneck to the ancient population ofA.spicatum(Wang et al.,2015),which could be in favor of the stochastic fixation of the ancient genetic polymorphisms across successive speciation events due to genetic drift.The discordance between the cpDNA tree and the nuclear MP-EST tree seems to be a good example of this due to the random geographic distribution of such differential fixation,such as the genetic affinity between the geographically disjunctiveA.spicatumandA.farrerirather than the geographically adjacentA.spciatumandA.cyathophorum(Li et al.,2016).Such geographically disjunctive but genetic affinities in the cpDNA tree might suggest we should reject the chloroplast replacement hypothesis,in which the ancestorA.spicatumcaptured a chloroplast from the ancestorA.farreri,due to the lack of a second contact (Fig.S4).
4.2.Possibility of hemiplasy for the incongruent traits
Phylogeny-based ancestral trait state inference is an effective way to trace the trait evolution along the phylogeny,which provides a profound understanding to the phenotypic diversity in the nature.However,widespread GTD across the whole genome can recover different relationships among species,and even some unrelated species in a species tree are connected by some genes or regions.If trait changes occur in these discordant regions,a phylogeny-based inference of ancestral traits may produce a false pattern (Hahn and Nakhleh,2016; Guerrero and Hahn,2018; Wu et al.,2018).The evolution of the inflorescence has undoubtedly attracted our attention,especially in systematic and phylogenetic studies,because the enormous phenotypic diversity of the inflorescence is directly related to the reproductive success of plants.The conspicuously elongated spicate inAllium spicatumcompared to the umbel in congeners has been considered as an alternative strategy to adapt to the extreme alpine habitat(here represented by altitude),accompanied by consequential changes in seed size,flowering phase,and ovary size (Li et al.,2016,2019b).Our RASPbased ancestral state reconstruction showed the ancestral state of inflorescence at the internodes along the species tree of subg.Cyathophorawas most likely at the umbel state and distributed across the eastern QTP with high confidence(BS=100%)(Fig.4A),which was consistent with the previous results using nuclear or cpDNA gene trees (Li et al.,2016).This result indicated that tree topology seemingly does not affect estimates of the ancestral state of the inflorescence,although the spicate inflorescence ofA.spicatumhas variable phylogenetic position,resulting in a paraphyletically distributed umbel inflorescence in the phylogeny of subg.Cyathophora.Similar to the categorical traits,the quantitative traits also showed paraphyletic distributions for the specific trait values of all branch tips,although there was consistency among ancestral trait values of internal nodes.All of these trait-state inferences seem to support a random fixation of derived trait values from ancient states (Fig.4B).However,given the ~27%-38.9% GTD across the genomes of subg.Cyathophora,this however has inevitably led to hemiplasy(Degnan and Rosenberg,2009;Guerrero and Hahn,2018).
It has been documented that higher HRFs imply a higher probability of hemiplasy than homoplasy regarding the incongruent traits between closely related species.When the HRFs >0.95,hemiplasy could be ten times more likely than homoplasy,and when HRFs <0.50 homoplasy is up to ten times more likely than hemiplasy(Guerrero and Hahn,2018).When given a constant substitution rate per nucleotide site per year,our HRFs were found to decrease as effective population size increased (Fig.4).In our study,theNe(133,870,95%HPDI[6310,978,952])used to calculate spicate and umbel inflorescence internode was much larger than the currentNeofA.spicatum(9821,95%HPDI[3162,51,591]),which has a population bottleneck (the ancientNeis 334,95% HPDI [32,7986]) (Wang et al.,2015),and thus HRFs would reach approximately 1 in the context of a smallNefor the ancestorAllium spicatum,implying a higher probability of hemiplasy regarding the trait transition betweenA.spicatumandA.cyathophorum-A.farreri,and in contrast to a lower shared probability of derived alleles (Guerrero and Hahn,2018).In addition,we found two neutral genes correlated with inflorescence development that show discordant tree topologies with the nuclear MP-EST tree(Fig.4F and G).This may also give support for the occurrence of incongruent inflorescence in the small ancient population ofA.spicatumdue to the random fixation of the ancient polymorphisms.Of course,identifying the genes that directly determine the formation of spicate and umbel inflorescences would provide more robust evidence for this conclusion.In addition,we found that the HRF at the internode ofA.cyathophorumandA.farreri,was higher than that at the internode ofA.spicatumandA.cyathophorum-A.farreri(Fig.4C,D,E).This is because hemiplasy most likely underlies the character state transition in the presence of ILS,suggesting the relevant variation occurred in an earlier clade and the following successive lineages are randomly sorted from these ancient polymorphisms (Guerrero and Hahn,2018).
5.Conclusions
In this study,phylotranscriptomic analyses of the four diploid species ofAlliumsubg.Cyathophorarevealed widespread genomewide gene-tree discordance (GTD),as well as incongruence between plasmid and nuclear genomes.Our coalescent simulations supported that substantial intergenomic ILS could have caused such GTD.We found that in the presence of ILS,hemiplasy is more likely than homoplasy to explain the trait transition between the current consecutive lineages.This is due to differential fixation of ancestral segregating variants.Our study exemplifies how to convert such phylogenetic noise into evolutionary information.This will provide a deeper understanding of how character-state transitions drive speciation and diversification.
Author contributions
MJL designed research.MJL collected samples.MJL,ZZZ and GL analyzed data.MJL and ZZZ writing original draft.All the authors revised and agreed to the published version of the manuscript.
Data availability statement
The transcriptomes under PRJNA598096 and complete chloroplast genomes have been deposited in the National Center for biotechnology information(NCBI),with public accession at https://www.ncbi.nlm.nih.gov/biosample.Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We would like to thank Dr.Daniel Petticord at the University of Cornell for his assistance with English language and grammatical editing of the manuscript.This work was supported by the Key Science & Technology Project of Gansu Province (22ZD6NA007),the National Key Research and Development Program of China(2021YFD2200202).Computing support was provided by the Supercomputing Center of Lanzhou University.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pld.2023.07.004.

- 植物多樣性的其它文章
- Flower morphology of Allium (Amaryllidaceae) and its systematic significance
- How to fill the biodiversity data gap:Is it better to invest in fieldwork or curation?
- Life forms affect beta-diversity patterns of larch forests in China
- A phylogenetic approach identifies patterns of beta diversity and floristic subregions of the Qinghai-Tibet Plateau
- Community structure and species diversity dynamics of a subtropical evergreen broad-leaved forest in China: 2005 to 2020
- Conservation genomic investigation of an endangered conifer,Thuja sutchuenensis,reveals low genetic diversity but also low genetic load