
Oxide-supported metal catalysts for anaerobic NAD+ regeneration with concurrent hydrogen production
2024-04-05 02:28JinweiLiJosephBurnettCludiMrtinezMcisRussellHoweXiodongWng
Chinese Chemical Letters 2024年2期
Jinwei Li ,Joseph W.H.Burnett ,Cludi Mrtinez Mcis ,Russell F.Howe ,Xiodong Wng,*
a Chemical Engineering,School of Engineering,Lancaster University,Lancaster LA1 4YW,United Kingdom
b Chemistry Department,University of Aberdeen,Aberdeen AB24 3UE,United Kingdom
Keywords: NAD+ regeneration Heterogeneous catalyst Hydrogenase mimic Hydrogen binding energy Hydrogen production
ABSTRACT We report SiO2-supported monometallic Pt,Pd,Au,Ni,Cu and Co catalysts for proton-driven NAD+ regeneration,co-producing H2.All metals are fully selective to NAD+ where the order of turnover frequencies(Pt >Pd >Cu >Au,Ni and Co) coincides with those otherwise observed in electrochemical hydrogen evolution reactions.This has revealed that NADH is capable of converting the metal sites into a “cathode”without an external potential and the NADH to NAD+ reaction involves transferring electron and hydrogen atom separately.Electron-deficient Ptδ+(on CeO2) enhances TOF and the heterogeneous Pt/CeO2 catalyst is recyclable without losing any activity/selectivity.
Biocatalysis using enzymes has attracted increasing interest in recent decades due to its outstanding activity and chemo-,regioand stereo-selectivity in biosensor,pharmaceutical and organic synthesis [1,2].As most enzymes intrinsically work under benign conditions,they are also able to lower energy costs,avoid extreme pH requirements as well as reduce environmental hazards [3-8].Dehydrogenases (DHs) are a group of important enzymes that catalyze reversible oxidative transformations of hydroxyls and amines(Fig.1a),some of which are novel reaction routes over conventional organic synthesis.The usage of DHs is limited by their dependence on the NAD(P)H/NAD(P)+cofactor pair (Fig.1b),which act as an electron carrier and is consumed stoichiometrically in enzymatic redox reactions.Due to the high cost of these cofactors (e.g.,$1400 and $2600 per mol of NAD+and NADH,respectively) [9],a regeneration system is essential to make these biosynthetic processes viable (Fig.1c) [10,11].Compared to the commercial uses of NAD(P)H and its regeneration,less attention has been paid to the regeneration of NAD(P)+,hampering the application of DH-catalyzed oxidative transformations [12].
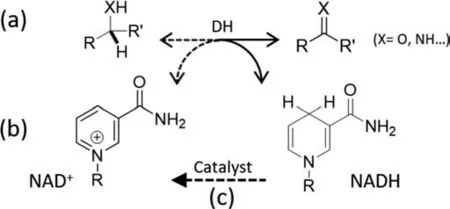
Fig.1.Schematic of dehydrogenases (DHs)-catalyzed oxidative biotransformations(a),molecular structure of cofactors (b) and catalytic NAD+ regeneration (c).Solid and dash arrows in (a) represent reversible oxidative and reductive transformations catalyzed by DHs,respectively.R: adenine dinucleotide.
To date,enzymatic NAD+regeneration using a second enzyme as the regeneration catalyst (e.g.,lactate dehydrogenases,glutamate dehydrogenases,and particularly,NADH oxidases (NOXs)) has been mostly studied [13-16].Although NOXs can generate H2O (from O2) as a clean product [15],the costliness,instability,and difficulty in downstream separation of enzymes remain challenging.New catalytic regeneration approaches based on homogeneous [17,18],electro-[19-21],and photo-catalysis [22-25] have been reported but are still at developmental stages.Improvements are required in regeneration efficiency (selectivity and activity),ease of preparation/separation,and system compatibility [21,26-28].
The use of solid catalysts for NAD+regeneration is a newly developed method where only supported Au,Pt and graphene oxide(Cu2+ion-doped) have so far been studied [29-31].The advantages of using supported metal catalysts lie in their low-cost preparation/setup,ease of separation,recyclability and scaling-up,and stable physicochemical properties.The previously reported catalytic systems were only active when using strong oxidant,molecular O2.We have recently established a new regeneration pathway employing activated carbon supported Pt that promoted NAD+regeneration using the readily available solution protons and concurrently produce H2(Eq.1) [32].Oxidizing NADH with a proton is more challenging as the latter is a significantly weaker oxidant than O2but it offers the possibility of avoiding the O2-liquid interfacial issues of deactivation [33-38] and has better potential for large scale applications with the further benefit of producing H2.
Carbon has been an excellent carrier for cofactor regeneration due to its own capability in electron transfer [39,40].However,this makes it difficult to decouple the effects of the support and the metal (i.e.,Pt,Pd,etc.) which hinders understanding of the role of the metal sites.In this work,we aim to address this gap of knowledge in proton-driven NAD(P)+regeneration.For the first time,a group of monometallic catalysts comprising Pt,Pd,Au,Ni,Cu or Co supported on non-conductive metal oxides (SiO2,MgO and CeO2)have been examined,with the role of metal revealed.Very surprisingly,with its capacity of donating electrons,NADH has effectively made a thermal/heterogeneous catalytic process “electrocatalytic”,governed by the hydrogen evolution reactions.
Full details of the materials used,catalyst preparation and characterization procedures are given in Supporting information.Catalysts were characterized by chemical analysis,X-ray powder diffraction,transmission electron microscopy,surface area and pore size analysis,and hydrogen temperature-programmed reduction.NAD+regeneration reactions were performed as described in Supporting information.The NADH concentration and conversion were determined by tracing the UV absorbance at 340 nm while the NAD+concentration and selectivity were monitored using enzymatic assays described in Supporting information and in our previous work [41].The reaction rate constants (k) were determined following first order kinetics.Turnover frequencies (TOF: NADH reacted per surface metal atom per hour) were calculated from the rate constants and the mean particle sizes obtained from TEM analysis,assuming spherical particles.The amount of hydrogen produced was measured volumetrically as described in Supporting information,and the gas composition confirmed by gas chromatography.
Table 1 summarizes the characterization data for the SBA-15 supported Pt,Pd,Au,Ni,Co and Cu catalysts tested for NAD+regeneration in this work.All catalysts showed similar distributions of metal particle sizes (TEM images in Fig.2) and mean particle diameters of around 4 nm,and nitrogen adsorption/desorption showed negligible pore blockage of the SBA-15 following addition of metal.
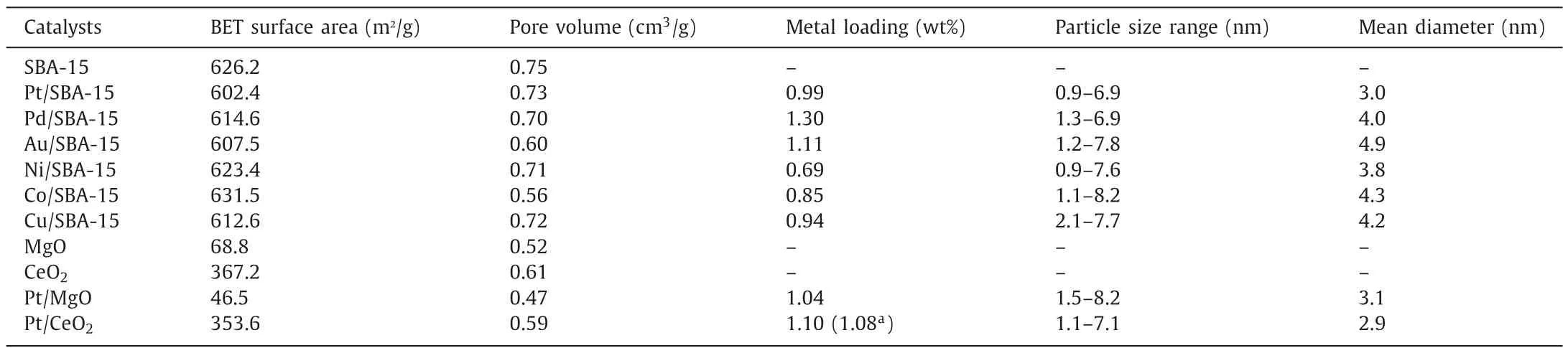
Table 1Catalyst characterization of all supported metal catalysts used.

Fig.2.Representative TEM images and size distribution histograms of the reduced catalysts: Pt (a),Pd (b),Au (c),Ni (d),Co (e) and Cu (f) on SBA-15.
The data shown in Fig.3a are NADH conversions after 1 h of reaction at pH 7.As illustrated in Fig.3a,only SBA-15 supported Pt,Pd and Cu give NADH conversions significantly higher than that seen in the absence of catalyst.The uncatalyzed decay of NADH is due to hydration of C=C bonds in the nicotinamide ring,which causes an increase in absorbance at around 290 nm and does not form NAD+[42,43].Fig.S4 (Supporting information)illustrates this (homogeneously) acid catalyzed undesirable reaction.Fig.3b shows time dependence of the UV-vis spectrum during NADH conversion over Pt/SBA-15 at pH 7.The decreasing absorbance at 340 nm due to the nicotinamide ring is not accompanied by any change at 290 nm,indicating that the undesired hydration reaction does not occur in the presence of the heterogeneous catalyst.Fig.3c plots NADH concentrationversustime at three different pHs and the corresponding first order plots are given in Fig.3d.Clearly,an acidic environment gave the highest conversion(i.e.,80% at pH 5,30% at pH 7 and 15% at pH 9).The obvious promoting effect of acidic conditions indicates the participation of H+in the regeneration reaction.A further regeneration test in DMSO,a non-proton polar solvent,was carried out in lieu of buffer solution.Since DMSO is incapable of providing H+,the NADH concentration remained unchanged after 1 h,showing the need of protons for the reaction to occur in contrast to the results obtained for the reaction in a phosphate buffer (Fig.S5 in Supporting information).Interestingly,a significant consumption (seen from 340 nm)was observed after an addition (1 mL) of concentrated phosphoric acid.These results further confirm the essential role of protons in the concerted NADH conversion,rather than a sequential step.Nevertheless,the peak rise at 290 nm was also observed which is an indicator of NADH decay product.This process was inevitable as the concentrated acid also contained water resulting in the hydration of NADH.However,this effect was inconsequential as it happens at a lower rate than that of the catalytic NAD+regeneration (Fig.3d and Fig.S4c).The first order kinetics demonstrated here is contrast with the second order kinetics reported for the oxygen-assisted NADH oxidation over platinum nanoparticles [31],suggesting a different reaction mechanism is operating.The selectivity of the NADH conversion to NAD+was determined by enzymatic assay of the NAD+concentration.For example,converting 0.255 mmol/L of NADH over Pt/SBA-15 at pH 7 for 1 h (Fig.3a) produced 0.250 mmol/L of NAD+,indicating close to 100% selectivity.
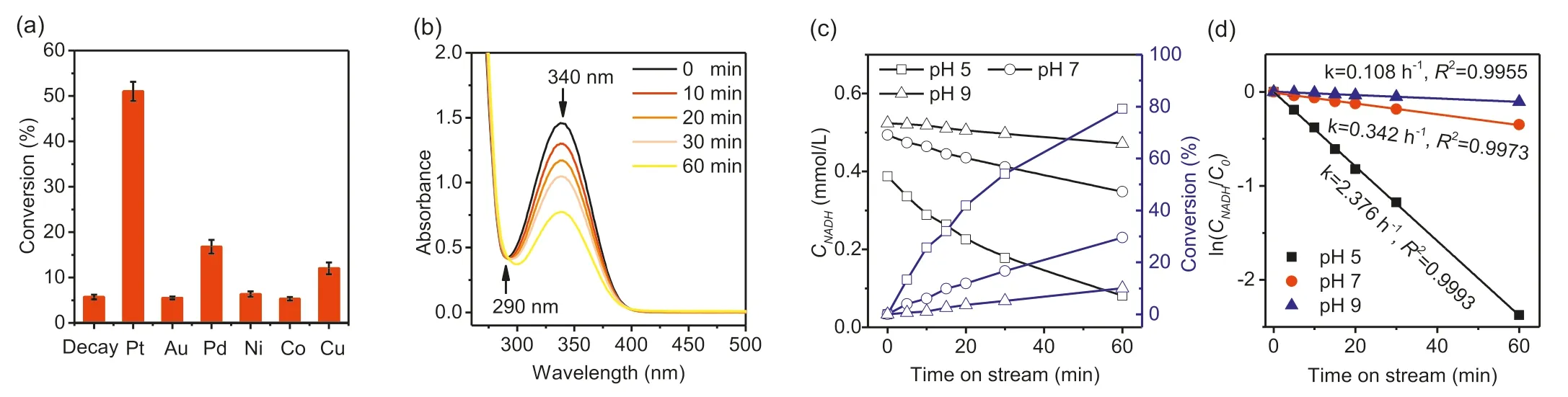
Fig.3.NAD+ regeneration promoted by the SBA-15 supported metal catalysts: NADH conversions over different metals (a),UV-vis spectrum scans of NAD+ regeneration over Pt/SBA-15 (b),temporal NADH concentration and conversion at different pH over Pt/SBA-15 (c) and first order kinetic plots (d).Reaction conditions: 37°C,0.5 mmol/L NADH,pH 7 (for a and b),100 mL/min N2 flow,1 h,60 mg (a,b) or 30 mg (c,d) of catalyst.
In a separate (scaled up) experiment,the production of hydrogen during the reaction was measured.After 12 h of reaction at pH 7 with 120 mg of catalyst and an initial NADH concentration of 2 mmol/L,the amount of hydrogen produced (4.2 mL) was close to that expected (4.8 mL) for the observed NADH conversion of 2 mmol/L and the reaction stoichiometry in Eq.1.Thus,despite the weaker oxidation potential than O2(0.82 V forE0(O2/H2O)vs.-0.41 V forE0(H+/H2),at pH 7) [44],H+was able to drive the regeneration in the presence of Pt.Opposed to the existing O2-assisted attempts on supported Au and Pt,the catalyst in this work functionally mimicked (NAD(H)-linked) hydrogenases which catalyze the conversion of NADH to NAD+and H2(Eq.1).
Two possible scenarios for the mechanism of the reaction in Eq.1 are illustrated in Fig.4.The first (Fig.4a) involves a direct hydride transfer to a proton to form H2at the platinum,while the second (Fig.4b) is an indirect hydride transfer and involves the electron and the extracted/adsorbed hydrogen atom being transferred separately to a proton to form hydrogen.The observed sequence of activities of the different metals supported on SBA-15(Pt>>Pd>Cu>Ni,Co,Au) is in excellent agreement with that of the electrochemical hydrogen evolution reaction (HER) in which electrons are combined with protons at a cathode to form hydrogen,viaeither the Volmer-Heyrovsky (Eqs.2 and 3) or the Volmer-Tafel (Eqs.2 and 4) sequences [45-48].

Fig.4.Plausible reaction pathway of NAD+ regeneration over a metal surface.The gray rectangle and star (*) represent metal particle and its surface site,respectively.
The hydrogen binding energy (HBE) has been well established for correlating with the HER activity of metals [45-48].The high HER activity of platinum is attributed to the optimum hydrogen binding affinity as revealed in the volcano plots (Fig.S6 in Supporting information).NADH is a well-known electron donor in nature.When it is continuously supplied/activated on a metal site such as platinum,the surface environment may be considered as mimicking an electrochemical HER at a cathode.Given the excellent agreement in the trends of metal activity between the two types of reaction,it may be argued that a mechanism for NAD+regeneration involving steps equivalent to the HER (Fig.4b) is plausible.The metal acts as a reservoir to accommodate hydrogen atoms (Had)and transfer electrons.Concertedly,an electron from NADH combines with a proton from the solution which then reacts with Hadto release H2(i.e.,the Volmer-Heyrovsky process (Eqs.2 and 3).Alternatively,two Hadcan also react to form H2(i.e.,the Volmer-Tafel process (Eqs.2 and 4).The key is that both processes rely on Hadinstead of hydride as an intermediate.Indeed,Attardetal.claimed that the formation of hydride on Pt (i.e.,Fig.4a) is thermodynamically unfavorable in aqueous solutions,further supporting the above analysis [49].The rate determining step of this NADH to NAD+conversion is therefore the HER,as opposed to NADH adsorption/activation or NAD+desorption.
The electronic properties of oxide supported platinum catalysts are known to be influenced by the nature of the support [50,51].In Fig.5a,we have compared the conversion of NADH under the same reaction conditions over SBA-15,CeO2and MgO supported platinum.All three catalysts contain similar platinum loadings and the mean particle sizes,and particle size distributions are closely similar (see TEM analyses in Figs.S7c and d in Supporting information),ruling out any effect of particle size on the activity.Additionally,the effect of the oxide supports (coming from different porous structure) was also discarded by examining NADH conversion over bare supports,as shown in Fig.S8 (Supporting information).The TOFs calculated from first order plots are respectively 39,30 and 9 h-1for Pt supported on CeO2,SBA-15 and MgO.All three catalysts were completely selective to NAD+as determined by enzymatic assays.XPS spectra in the 4f region (Fig.5b) show that platinum on CeO2was electron deficient relative to metallic platinum black (Pt 4f7/2binding energy of 72.5 eV compared with 71.6 eV for platinum(0),Fig.S9 in Supporting information),whereas platinum on SBA-15 has a 4f7/2binding energy of 71.6 eV and platinum on MgO 71.4 eV.We suggested that the electron deficient nature of the platinum on CeO2led to a reduced hydrogen binding strength,favoring the desorption of hydrogen.Fig.5c shows the correlation between TOF and Pt 4f7/2binding energy.A similar conclusion has been reported concerning the advantage of electron deficient platinum for HER kinetics [52-54].
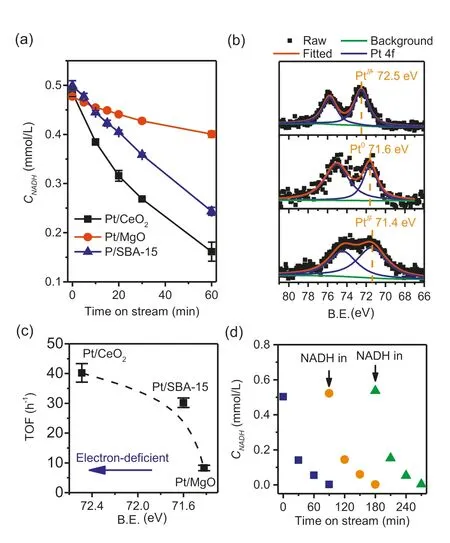
Fig.5.NADH conversion over Pt/CeO2,Pt/MgO and Pt/SBA-15 catalysts (a),Pt 4f XPS spectra of the catalysts (b),correlation of TOF with binding energy (c) and catalyst recycle test with Pt/CeO2 (d).Reaction conditions: 0.5 mmol/L NADH,100 mL 0.1 mol/L pH 7 PBS,37°C,700 rpm stir,100 mL/min N2,60 (for a) or 120 mg (for d)catalyst.Error bars obtained from three independent experiments.
Finally,we tested the recyclability of the heterogeneous supported Pt catalyst using the most active Pt/CeO2as representative.To reach a full conversion,double amount of catalyst (120 mg) was used,and reactions were run with three fresh batches of NADH feed.Results are shown in Fig.5d.After the first run,a conversion of 100% was reached in 1.5 h and the doubled initial reaction rate (0.019 mmol L-1h-1) compared to 0.009 mmol L-1h-1(Fig.5a)confirmed that the reactions were still operating under kinetic control region (i.e.,catalyst was not in excess).The unchanged performance in the three successive reactions proved that there was no catalyst deactivation (e.g.,leaching,fouling or poisoning).In addition,ICP analysis of the spent catalyst shows that the Pt loading was unchanged (1.10vs.1.08 wt%) before and after reactions(Table 1).
For the first time,we proved the feasibility of H2productionviaH+-driven NAD+regeneration catalyzed by oxide-supported metal catalysts.Pt was shown as the most promising active phase among tested metals (Au,Pd,Ni,Cu and Co,all supported on SBA-15),where NADH was converted to NAD+with full selectivity.The trend of metal activity was found following the well-established HER volcano pattern correlated with hydrogen binding energy,confirming a hydrogen-electron transfer mechanism (as opposed to hydride transfer) regarding the hydrogen formation.Electronic structure of the Pt nanoparticles (tuned by using SBA-15,MgO and CeO2) was important for catalysis where electron-deficient Pt on nonconductive metal oxides favors the activity.This may be due to the lowered hydrogen binding strength with Pt.The results shed light on the design of heterogeneous catalytic systems for protondriven NAD+regeneration as well as a potential route for H2production by bio-oxidation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the EPSRC New Horizons grants(Nos.EP/V048635/1 and EP/X018172/1).We are also grateful for support from the UK Catalysis Hub funded by EPSRC grant reference EP/R026645/1.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108737.
 Chinese Chemical Letters2024年2期
Chinese Chemical Letters2024年2期
- Chinese Chemical Letters的其它文章
- The 3rd Xihua Chemistry and Biomedicine Forum
- Professor Hualiang Jiang: A tribute to an esteemed visionary chemist and pharmacist
- Recent advances in visible light-mediated chemical transformations of enaminones
- Development of porphyrin-based fluorescent sensors and sensor arrays for saccharide recognition
- Recent advances of versatile reagents as controllable building blocks in organic synthesis
- Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting☆