
Efficacy and Safety of lxekizumab in Chinese Patients With Moderate-to-Severe Plaque Psoriasis: 60-Week Results From a Phase 3 Study
2023-01-12 12:29XiaLiJieZhengWeiLiPanMinZhengYanLuFuQiuLiYangFengDingJianZhongZhangHongYingLiWenLongRui
國際皮膚性病學雜志 2022年4期
Xia Li, Jie Zheng,*, Wei-Li Pan, Min Zheng, Yan Lu, Fu-Qiu Li, Yang-Feng Ding, Jian-Zhong Zhang,Hong-Ying Li, Wen-Long Rui
1 Department of Dermatology, Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200025,China; 2 Department of Dermatology, Zhejiang Provincial People’s Hospital, Hangzhou, Zhejiang 314408, China; 3 Department of Dermatology, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang 310003, China;4 Department of Dermatology, Jiangsu Province Hospital, Nanjing, Jiangsu 210029, China; 5 Department of Dermatology, The Second Hospital of Jilin University, Changchun, Jilin 130041, China; 6 Department of Dermatology, Shanghai Skin Disease Hospital,Shanghai 130041, China; 7 Department of Dermatology, Peking University People’s Hospital, Beijing 130041, China; 8 China Drug Development and Medical Affairs Center, Eli Lilly and Company, Shanghai 200041, China.
Abstract Objective: Ixekizumab is a high-affinity monoclonal antibody that selectively targets interleukin-17A and is approved for treating moderate-to-severe psoriasis. This phase 3, multicenter, randomized, double-blind, placebocontrolled trial (NCT03364309; registered December 6, 2017) evaluated the safety and efficacy of ixekizumab in Chinese patients with moderate-to-severe psoriasis.Methods: 438 patients were randomized 2:2:1 to 80 mg ixekizumab every 2 weeks (IXE Q2W, n = 176), 80 mg ixekizumab every 4 weeks (IXE Q4W, n = 174), or placebo (n = 88). Efficacy was assessed by evaluating the static Physician’s Global Assessment score of 0 or 1 (sPGA [0,1]) and Psoriasis Area and Severity Index (PASI) 75/90/100 responses, and nonresponder imputation was used for handling missing data. The safety profile was evaluated by assessing treatment emergent adverse events (AEs) and serious AEs.Results: At week 12, the sPGA (0,1) response rates were 3.4%, 79.9%, and 86.4% in the placebo, IXE Q4W, and IXE Q2W groups, respectively. The PASI 75/90/100 response rates were 8.0%/2.3%/0.0%, 87.4%/75.9%/29.3%,and 93.8%/82.4%/33.0% in the placebo, IXE Q4W, and IXE Q2W groups, respectively. Ixekizumab led to rapid PASI 50 responses, as early as week 1, whereas PASI 75 and sPGA (0,1) responses were observed from week 2. sPGA(0,1) and sPGA (0) responses were maintained through week 60 in a higher proportion of patients receiving IXE Q4W vs. placebo. The safety profile was consistent with previous studies of ixekizumab in psoriasis.Conclusion: Ixekizumab showed a rapid onset of action and high efficacy that was maintained through 60 weeks and was well tolerated with no unexpected AEs, in Chinese patients with moderate-to-severe plaque psoriasis.
Keywords: China, efficacy, ixekizumab, psoriasis, rapid onset, safety
lntroduction
Psoriasis is a life-long, chronic, inflammatory skin condition. In addition to characteristic skin plaques, psoriasis is frequently accompanied by various comorbidities including cardiovascular disease, psoriatic arthritis, impaired quality of life (QoL), and associated depressive illness.1Psoriasis can also adversely impact work productivity2and lead to sleep disturbances.3Hence, treating the manifestations of psoriasis is important and typically involves systemic therapy in patients with moderate-to-severe disease.
Recent estimates suggest that 60 million people worldwide have psoriasis, with the prevalence ranging from 0.14% in East Asia to 1.99% in Australasia.4The disease burden of psoriasis in China appears to be increasing with estimated prevalence ranging from 0.17% in 19845to 0.47% in 2012.6Approximately 97% of Chinese patients with psoriasis have plaque psoriasis,5hereafter referred to as psoriasis.
Important unmet needs in the treatment of psoriasis include the development of treatments with a rapid onset of action that result in complete clearance of skin lesions.This is supported by several studies showing that patients desire treatments with rapid efficacy that resolve all skin lesions.6-7Furthermore, patients achieving rapid clinical responses have been shown to experience greater cumulative clinical benefits, as well as better patient outcomes,including QoL, compared to those who achieve slower responses.8-9
Ixekizumab is a high-affinity immunoglobulin G4 monoclonal antibody that selectively inhibits interleukin (IL)-17A, an important cytokine mediator of psoriasis.12The results of 3 phase 3, global pivotal trials,UNCOVER-1, UNCOVER-2, and UNCOVER-3,1,10revealed that ixekizumab showed rapid onset and superior efficacy compared to etanercept and placebo and an established and consistent safety profile (total ixekizumab exposure, 3,458.4 patient-years) in patients with moderate-to-severe psoriasis. In China, ixekizumab has received approval for moderate-to-severe psoriasis and is included in the Chinese Society of Dermatology guidelines as a recommended biologic treatment.11However, the overall use of biologics in China for the treatment of psoriasis remains relatively low, and despite the approval of ixekizumab in China, there is a lack of data on the efficacy and safety of ixekizumab in Chinese patients.
Here, we report the 60-week efficacy and safety findings from a phase 3, randomized controlled trial of ixekizumab in Chinese patients with moderate-to-severe psoriasis.
Materials and methods
Study design
This was a multicenter, phase 3, randomized, double-blind,placebo-controlled, parallel-group trial conducted at 17 sites in China from April 2018 to June 2020 to investigate the effect of 2 ixekizumab dose regimens in Chinese patients with moderate-to-severe psoriasis. The study protocol was approved by the local independent ethics review boards with Approval ID (2017)LunShenDi(81)-4,and study conduct followed the principles outlined in the Declaration of Helsinki, the International Council for Harmonisation (Guideline for Good Clinical Practice),and applicable local laws and regulations. Written informed consent was obtained from all patients before inclusion, and the study was registered at clinicaltrials.gov (NCT03364309).
The study included a screening period, an induction dosing period (12 weeks), a rerandomized maintenance dosing period (48 weeks), and a posttreatment follow-up period. Results from the 12-week induction dosing period and the 48-week maintenance dosing period are reported here.
Study population
Eligible patients were aged ≥18 years and had chronic psoriasis for ≥6 months, a Psoriasis Area and Severity Index(PASI) score ≥12, a static Physician’s Global Assessment(sPGA) score of ≥3, and ≥10% psoriasis-affected body surface area at screening and baseline and were eligible for systemic therapy and/or phototherapy. Patients were excluded from the study if they had other forms of psoriasis (pustular, erythrodermic, and/or guttate forms), a history of drug-induced psoriasis, previous exposure to ixekizumab or any other biologic directly targeting IL-17 or the IL-17 receptor, or current or recent use of any biologic within the following times from baseline: infliximab,adalimumab, or alefacept <60 days; etanercept <28 days;ustekinumab <8 months; golimumab <90 days; rituximab or efalizumab <12 months; or any other biologic agent <5 half-lives.
Treatment protocol
Induction dosing period
At the beginning of the induction dosing period, patients were randomized 2:2:1 to receive ixekizumab 80 mg by subcutaneous injection every 2 weeks (IXE Q2W) after a starting dose of 160 mg (week 0) or ixekizumab 80 mg every 4 weeks (IXE Q4W) after a starting dose of 160 mg(week 0) or matched placebo.
Maintenance dosing period
Patients who completed the 12-week induction dosing period were entered into the rerandomized maintenance dosing period (weeks 12–60). At the beginning of this period, patients classified as ixekizumab responders(sPGA score of 0 or 1 [0,1]) during the induction dosing period were rerandomized (2:1) to IXE Q4W or placebo,with stratification by induction dosing regimen. Patients rerandomized to IXE Q4W received subcutaneous injections (one each) of ixekizumab and placebo at week 12 and IXE Q4W thereafter. Patients rerandomized to placebo received 2 subcutaneous injections of placebo at week 12 and placebo Q4W thereafter. Patients classified as placebo responders during the induction dosing period continued to receive placebo until relapse (sPGA score ≥3)and received IXE Q4W thereafter. Nonresponders who received any investigational product during the induction dosing period were assigned to IXE Q4W. Here we report the results for ixekizumab responders at the end of the induction dosing period who were rerandomized in the maintenance dosing period (ie, relevant treatment groups were IXE Q4W/placebo, IXE Q4W/IXE Q4W, IXE Q2W/placebo, and IXE Q2W/IXE Q4W).
Outcome measures
We evaluated the superiority of IXE Q2W or IXE Q4W to placebo for 2 coprimary end points at week 12: the proportion of patients achieving a ≥75% improvement in PASI (PASI 75) and sPGA (0,1). Major secondary end points evaluated during the induction dosing period included the proportion of patients achieving sPGA (0), ≥90% improvement in PASI (PASI 90), and 100% improvement in PASI (PASI 100); the change from baseline in the Nail Psoriasis Severity Index (NAPSI) in patients with baseline fingernail involvement; the proportion of patients with a ≥4-point reduction in the Itch Numeric Rating Scale (NRS) in patients with Itch NRS≥4 points at baseline; and the change from baseline in Dermatology Life Quality Index (DLQI) at week 12. In addition, the time course of PASI 50 was evaluated.
Major secondary end points during the maintenance dosing period included the proportion of patients maintaining or achieving sPGA (0,1) and sPGA (0) from week 12 after rerandomization to week 60. Additional secondary end points assessed during the maintenance dosing period included the proportion of patients achieving PASI 75, PASI 90, and PASI 100.
Safety was evaluated in both study periods by recording treatment emergent adverse events (TEAEs), including the severity and the relationship to study treatment,serious adverse events (SAEs), discontinuations or dose modifications owing to adverse events (AEs), and clinical laboratory measurements. AEs of special interest included liver function test changes/enzyme elevations, cytopenias,injection site reactions, infections, inflammatory bowel disease (IBD; including Crohn’s disease and ulcerative colitis), allergic reactions/hypersensitivities, malignancies,cerebrocardiovascular events (including major adverse cerebrocardiovascular events), depression, and interstitial lung disease.
Blinding
This was a double-blind study; both patients and study personnel were blinded to treatment allocation until the final database lock, after all patients had completed or discontinued from the study. Sponsors were unblinded after the database was validated and locked. Assignment to treatment was implemented using a computer-generated random sequence and an interactive web response system. Study personnel confirmed they received the correct investigational product package via a confirmation number. Study blinding was preserved by requiring the randomization table, and treatment assignments were seen by a minimum number of sponsor personnel, who were not directly in contact with the study sites. Blinding during the induction dosing period was maintained by giving all patients 2 injections at week 0 and 1 injection every 2 weeks from week 2 through 10. Blinding during the maintenance dosing period was maintained by giving all patients 2 injections at week 12 and 1 injection every 4 weeks from week 16 through 56.
Statistical analysis
The planned sample size was 420 patients. Assuming sPGA(0,1) and PASI 75 response rates at week 12 of 75% for each ixekizumab treatment group and 5% for placebo,this sample size provided >99% power to test the superiority of each ixekizumab dose regimenvs. placebo for the coprimary end points at an α of 0.025 using a 2-sided Fisher exact test. These assumptions were based on the integrated results of the pivotal phase 3 psoriasis studies,UNCOVER-1, UNCOVER-2, and UNCOVER-3.1,12
Efficacy analyses for the induction dosing period were conducted on an intention-to-treat population,defined as all randomized patients, regardless of whether the patient received the assigned treatment.Efficacy analyses during maintenance period were conducted in a maintenance dosing period primary population (all patients randomized to ixekizumab during the induction dosing period achieving sPGA [0,1] at week 12 were rerandomized and then received ≥1 dose of study treatment during the maintenance period).Categorical outcomes were analyzed with logistic regression, taking treatment as a covariate, and missing data were imputed using nonresponder imputation.Patients who did not meet the clinical response criteria or had missing clinical response data at the analysis time point were considered nonresponders in the nonresponder imputation analysis. In cases where a logistic regression model did not produce results owing to sparse data, the Fisher exact test was used. All continuous outcomes were analyzed using a mixed-effects model for repeated measures with the following fixed factors: treatment, baseline value, visit, treatment-byvisit interaction, and baseline-by-visit interaction. A gatekeeping strategy was implemented for the primary and major secondary analyses to control the studywise 2-sided α at 0.05 (see the Supplemental Online Resources, http://links.lww.com/JD9/A29, for further details). The underlying procedure was derived using the methodology developed by Dmitrienko and Tamhane.12There was no adjustment for multiple comparisons for any other analyses.
Safety analyses for the induction dosing period were conducted in all randomized patients who received ≥1 dose of ixekizumab (safety population), while safety analyses for the maintenance dosing period were conducted on the maintenance dosing period primary population.
All analyses were performed using SAS, version 9.4(Cary, NC).
Corona virus disease 2019 (Covid-19)pandemic–related amendments to statistical analysis
The COVID-19 pandemic occurred after the trial was fully enrolled. All 17 sites were impacted by the pandemic, and 15 sites returned to normal before database lock occurred in August 2020. To ensure participants’safety, study quality, and integrity, remote visits (phone calls or virtual visits) were used to replace protocol-required on-site visits if on-site visits were not feasible owing to traffic restrictions and/or safety concerns during the pandemic. Where protocol assessments could not be done remotely (eg, physician-reported questionnaires, vital signs, electrocardiography, laboratory tests), data were considered missing. Remote visits that were out of the visit window were reported as protocol deviations.
In response to the COVID-19 pandemic, the statistical analysis plan was amended to include listings of participant disposition from study/study treatment due to COVID-19 and protocol deviations due to COVID-19. A post hoc analysis was conducted on the Maintenance Dosing Period Primary Population to evaluate the impact of missing data owing to COVID-19 on PASI 75/90/100, sPGA (0,1) and sPGA (0) responses at weeks 52, 56, and 60.
Results
Patient disposition, demographics, and baseline clinical characteristics
A total of 438 patients were randomly assigned to receive placebo (n= 88), IXE Q4W (n= 174), or IXE Q2W(n= 176). Across all treatment groups, 427 (97%) patients completed the 12-week assessment; a similar number of patients in each treatment group discontinued treatment in the induction period (Fig. 1A).
Among 350 patients receiving ixekizumab in the induction period, 291 were classified as responders at week 12,and 289 (139 from the IXE Q4W group and 150 from the IXE Q2W group) were rerandomized to placebo or IXE Q4W (Fig. 1B). Of the patients rerandomized to ixekizumab, 164 (85%) completed the maintenance dosing period without relapse, whereas 88 (91%) patients rerandomized to placebo relapsed (ie, sPGA ≥ 3).
Baseline characteristics were well balanced across the treatment groups (Table 1). Among the overall intention-to-treat population (n= 438), the mean (SD) age was 40.5 (11.4) years, and 76.5% of the patients were male. The mean (SD) body weight was 72.9 (13.9) kg,disease duration was 16.1 (8.9) years, baseline PASI score was 25.6 (10.1), and percentage body surface area involvement was 42.5% (21.2%). Overall, 74.2% of patients had received previous nonbiological systemic therapy and 9.4% had received previous biological therapy.
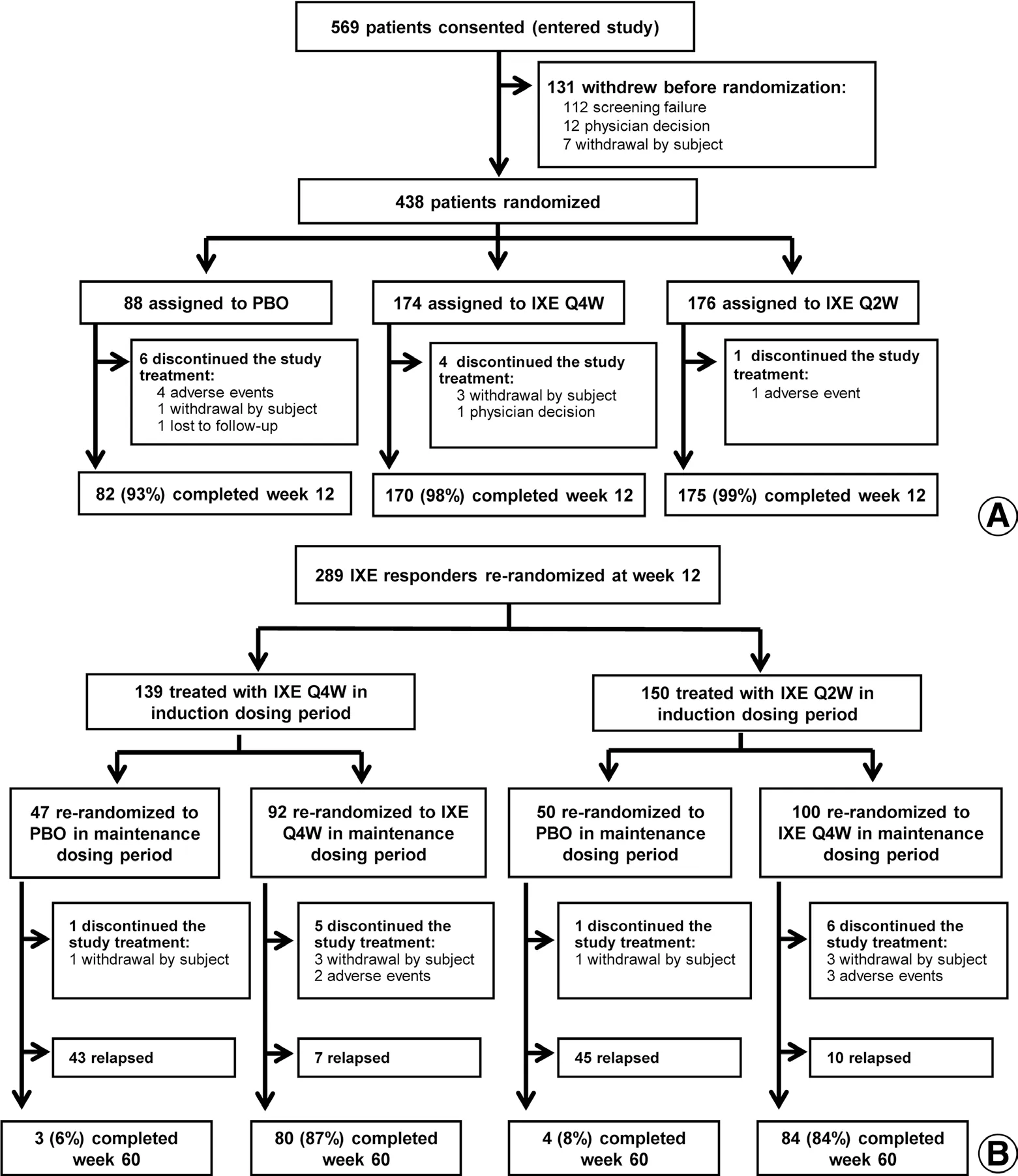
Figure 1. Patient flowchart. (A) Induction dosing period. (B) Maintenance dosing period. IXE: ixekizumab; IXE Q2W: 80 mg IXE every 2 weeks;IXE Q4W: 80 mg IXE every 4 weeks; PBO: placebo.
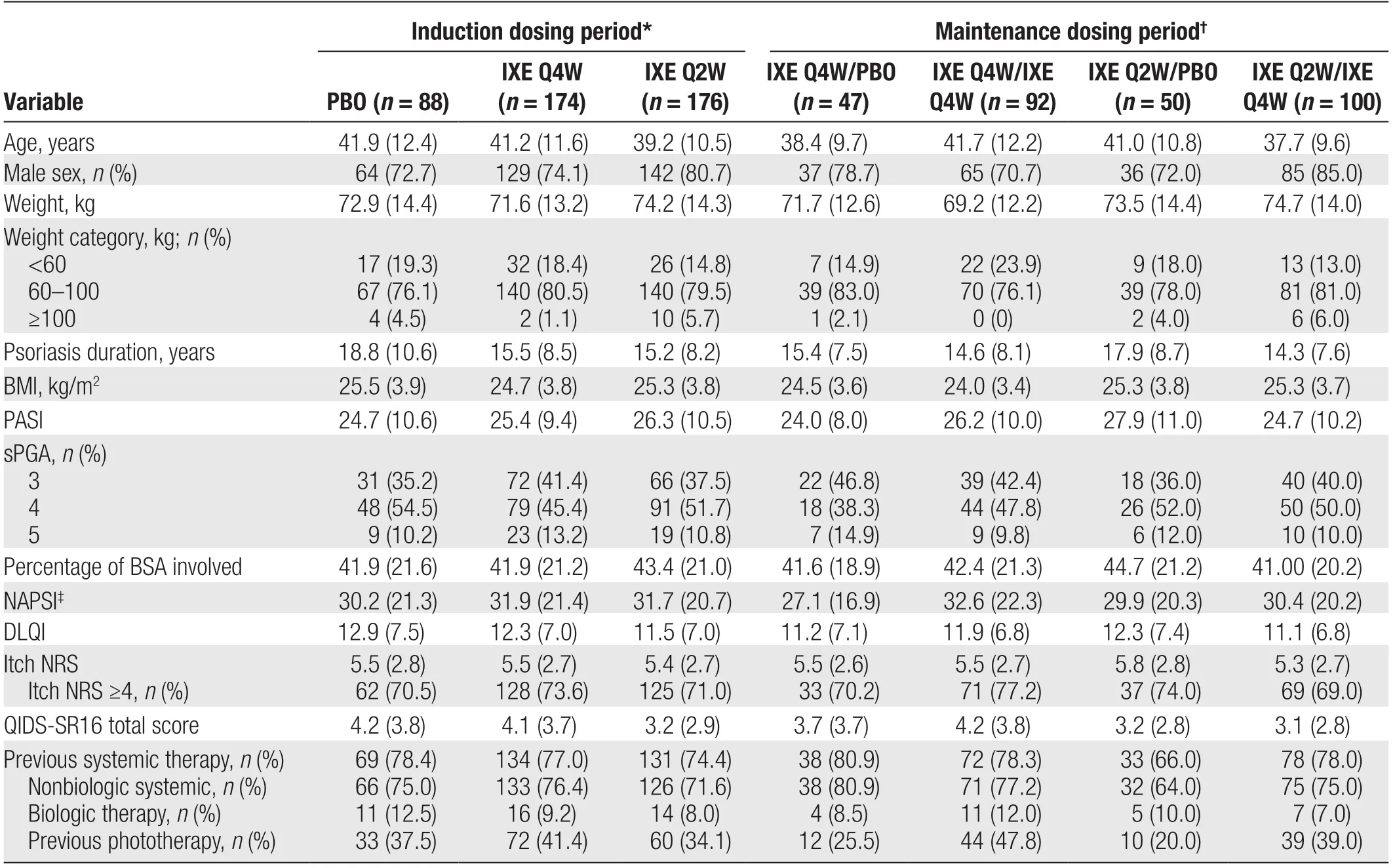
Table 1 Baseline demographics and clinical characteristics.
Efficacy outcomes
Induction dosing period
All coprimary end points and major secondary end points were met and showed greater improvements with both ixekizumab regimensvs. placebo (Table 2). For the coprimary end points PASI 75 and sPGA (0,1), significantly greater efficacy was observed with both ixekizumab regimens than with placebo at week 12 (allP< 0.001;Fig. 2A and 2C; Table 2). In addition, a higher proportion of patients in the IXE Q4W and IXE Q2W groups achieved PASI 75 at week 12 (87.4% and 93.8%vs.8.0%;P< 0.001) and sPGA (0,1) at week 12 (79.9%and 86.4%vs. 3.4%;P< 0.001)vs. the placebo group,respectively (Fig. 2A and 2C).
All major secondary end points were met; and significantly greater improvements in PASI 90, PASI 100, and sPGA (0) response rates at week 12 were observed with both ixekizumab regimensvs. placebo (allP< 0.001;Fig. 2B, 2D, and 2E; Table 2). Among patients in the IXE Q2W group, 82.4% had attained near-complete resolution (PASI 90) and 36.4% had attained complete resolution (sPGA [0]) at week 12. A significantly greater proportion of patients in both ixekizumab groups achieved sPGA (0) and PASI 90 as early as week 4vs. placebo (allP< 0.01; Fig. 2B and 2D). In patients with nail psoriasis, those receiving IXE Q4W and IXE Q2W had significantly greater reductions from baseline in NAPSI at week 12 compared with patients receiving placebo (bothP< 0.001; Fig. 2F; Table 2).
In patients with an Itch NRS score of ≥4 at baseline, significant differences in the proportions of patients achieving a ≥4-point reduction in Itch NRS were observed as early as week 1 in both ixekizumab groupsvs. placebo and were maintained through week 12 (P< 0.01 at week 1;P< 0.001 at week 12; Fig. 3A;Table 2). Quality of life (QoL), as measured by the change from baseline in DLQI, improved as early as week 2 in both ixekizumab groups and was maintained through week 12 (P< 0.001vs. placebo at weeks 2 and 12; Fig. 3B; Table 2).
Treatment with ixekizumab was associated with early onset of efficacy. Patients in the IXE Q4W and IXE Q2W groups achieved significantly higher PASI 50 response rates compared with those in the placebo group as early as week 1 (32.2% and 28.4%vs. 3.4%; bothP< 0.001;Supplemental Online Resource 1, http://links.lww.com/JD9/A29). Similarly, response rates for PASI 75 and sPGA(0,1) were significantly higher in the IXE Q4W and IXE Q2W groupsvs. the placebo group from week 2 (PASI 75:24.1% and 21.0%vs. 0%; bothP< 0.001; Fig. 2C; sPGA(0,1): 21.3% and 17.6%vs. 0%; bothP< 0.001; Fig. 2A).
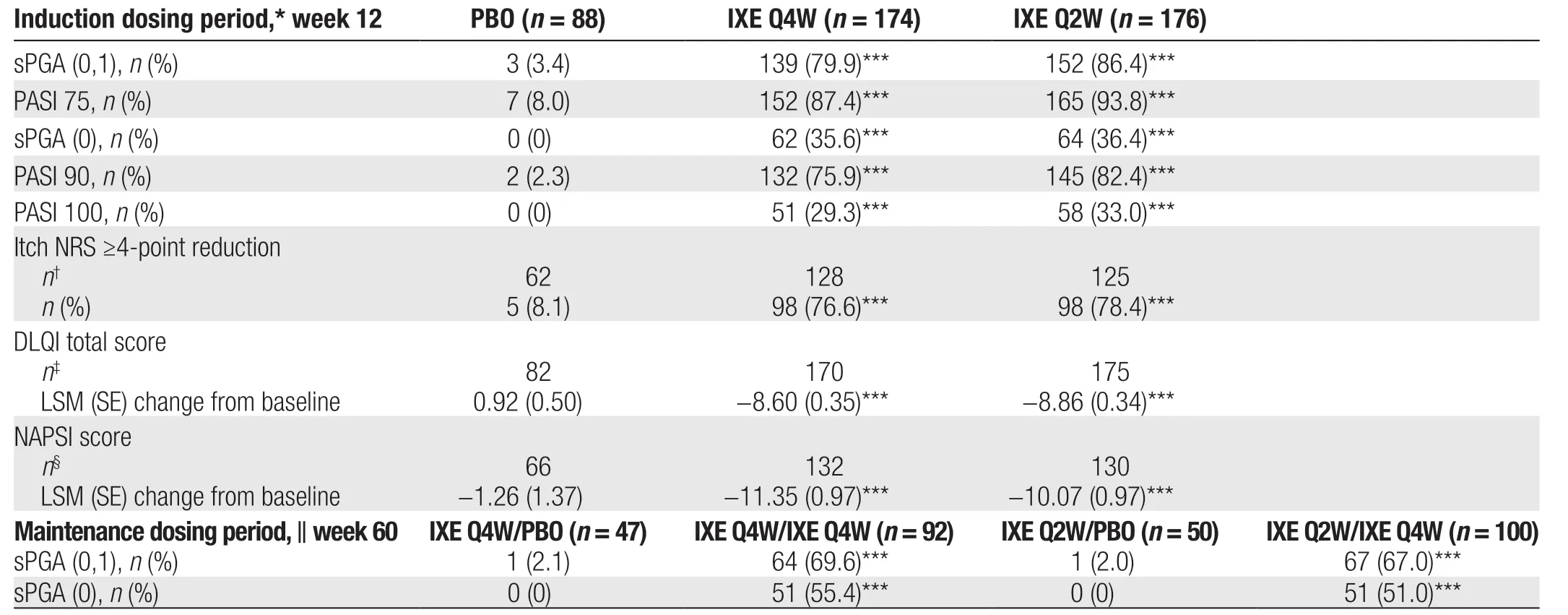
Table 2 Coprimary end points and all major secondary end points.
Maintenance dosing period
Ixekizumab sustained high efficacy during the maintenance dosing period from weeks 12 to 60 after rerandomization (Supplemental Online Resource 2, http://links.lww.com/JD9/A29). At week 60, there were significantly greater improvements in sPGA (0,1) (bothP< 0.001;Table 2; Supplemental Online Resource 2A, http://links.lww.com/JD9/A29) and sPGA (0) (bothP< 0.001; Table 2;Supplemental Online Resource 2B, http://links.lww.com/JD9/A29) with both ixekizumab regimensvs. placebo.
Impact of COVID-19 pandemic on efficacy results
Owing to the COVID-19 pandemic, 34 patients had remote visits at week 60 and thus had missing efficacy measures in the maintenance period primary population.When these patients were excluded, PASI 75 and sPGA(0,1) response rates were higher at week 60 in all ixekizumab treatment groups compared with responses when these patients were not excluded (overall for patients who received ixekizumab during the induction and maintenance dosing periods: 76.6%vs. 68.2% for sPGA [0,1]and 84.2%vs. 75.0% for PASI 75, respectively). Similar results were also seen for PASI 90, PASI 100, and sPGA (0)responses at week 60. Only 1 patient had a remote visit at week 52, and no patients had a remote visit at week 56 due to the pandemic. Thus, the impact of COVID-19 on the efficacy results at week 52 were minimal, and there was no impact on results at week 56.
Safety and tolerability measures
In the induction and maintenance dosing periods, the percentage of patients reporting TEAEs was higher in the ixekizumab treatment groups than in the placebo groups (Table 3). Rates of TEAEs were similar between the ixekizumab treatment groups in both dosing periods (Table 3). Among the patients receiving ixekizumab during the induction dosing period, the most common TEAEs included upper respiratory tract infection, injection site reaction, and tinea pedis (Table 3). Most TEAEs were mild or moderate in severity. SAEs were reported by ≤5.4% of patients across all treatment groups in the induction and maintenance dosing periods. The incidence of TEAEs leading to discontinuation was similar between all ixekizumab treatment groups in both dosing periods.
The rates of some AEs of special interest were higher in the ixekizumab treatment group than in the placebo group (Table 3). Infections and injection site reactions occurred more frequently in patients who received ixekizumab than those who received placebo in both the induction and maintenance dosing periods. All injection site reactions were mild to moderate in severity, and no patient discontinued owing to this AE. The most frequent infections (occurring in ≥5% of patients receiving ixekizumab) were upper respiratory tract infection and tinea pedis. Hepatic events were more common in the IXE Q4W group than in any other group. One patient in the IXE Q2W/IXE Q4W group discontinued the study in the maintenance dosing period owing to a hepatic event (mild hepatic function abnormality).

Figure 2. Time course of patient responses during the induction dosing period (weeks 0–12; placebo [PBO], n = 88; 80 mg ixekizumab every 4 weeks [IXE Q4W], n = 174; 80 mg ixekizumab every 2 weeks [IXE Q2W], n = 176). (A) Percentage of patients with static Physician’s Global Assessment (sPGA) of 0 (clear) or 1 (minimal psoriasis). (B) Percentage of patients with sPGA (0). (C) Percentage of patients with reduction from baseline in PASI ≥75% (PASI 75). (D) Percentage of patients with reduction from baseline in PASI ≥90% (PASI 90). (E) Percentage of patients with reduction from baseline in PASI of 100% (PASI 100). (F) Change from baseline in Nail Psoriasis Severity Index (NAPSI) for patients with nail involvement (PBO, n = 71; IXE Q4W, n = 133; IXE Q2W, n = 131; data are least squares mean ± standard error). Included patients in the intention-to-treat population; all randomized patients, regardless of whether the patient followed the treatment protocol.Missing data were imputed using nonresponder imputation, and logistic regression was used for sPGA (0,1), sPGA (0), PASI 75, PASI 90,and PASI 100, while mixed-effects model for repeated measures analysis was used for change from baseline in NAPSI. In cases where a logistic regression model did not produce results owing to sparse data, the Fisher exact test was used. PASI: Psoriasis Area and Severity Index. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. PBO.
There were no clinically meaningful changes in laboratory findings during either treatment period. No patients were infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or diagnosed with Covid-19 during the study.
Discussion
In this study, neutralization of IL-17A with ixekizumab demonstrated a rapid onset of action, high rates of skin clearance, and sustainable efficacy for up to 60 weeks in Chinese patients with moderate-to-severe psoriasis. Notably, during the first 12 weeks of this study, we found that patients receiving IXE Q4W or IXE Q2W demonstrated early and high levels of clinical response in their psoriasis compared with patients receiving placebo. Continued exposure to ixekizumab every 4 weeks for a further 48 weeks led to the maintenance of these high response rates; complete skin clearance (PASI 100)was maintained or achieved by at least 50% of patients.Further, safety findings were consistent with the established safety profile of ixekizumab in psoriasis. Overall,the response of Chinese patients with psoriasis to treatment with ixekizumab was consistent with the responses observed in the global phase 3 clinical trials.1,10
Ixekizumab demonstrated high efficacy in Chinese patients with moderate-to-severe psoriasis, with all coprimary and major secondary end points met under stringent adjustments for multiple testing to control the study-wise type I error level at 0.05. Notably, by week 12, >85% of patients who received IXE Q2W achieved clear/almost clear skin (sPGA [0,1]), 94% achieved clinically meaningful responses (PASI 75), and 82% achieved a high-level response (PASI 90). These findings are similar to those observed for the predominantly White patient populations who received IXE Q2W in the global UNCOVER-1, UNCOVER-2, and UNCOVER-3 studies,where sPGA (0,1) response rates were 82%, 83%, and 81%, respectively; PASI 75 response rates were 89%,90%, and 87%, respectively; and PASI 90 response rates were 71%, 71%, and 68%, respectively.1,10In addition,consistent with the disease severity findings, the results of this study showed that both ixekizumab regimens were associated with significant improvements in nail psoriasis (NAPSI) and QoL (Itch NRS and DLQI) at week 12 compared with placebo.

Figure 3. Time course of patient responses during the induction dosing period (weeks 0–12). (A) Percentage of patients with≥4-point improvement in Itch numeric rating scale (NRS; placebo[PBO], n = 62; 80 mg ixekizumab every 4 weeks [IXE Q4W], n = 128;80 mg ixekizumab every 2 weeks [IXE Q2W], n = 125; missing data were imputed using nonresponder imputation; logistic regression).(B) Change from baseline in Dermatology Life Quality Index (DLQI)total score (PBO, n = 86; IXE Q4W, n = 174; IXE Q2W, n = 175;data are least squares mean ± standard error; mixed-effects model for repeated measures analysis). Included patients in the intentionto-treat population; all randomized patients, regardless of whether the patient followed the treatment protocol. sPGA: static Physician’s Global Assessment. ** P < 0.01, *** P < 0.001 vs. PBO.
The high rates of clinical improvement during induction dosing period were sustained throughout the maintenance dosing period in most patients. In general, patients received ixekizumab in the induction period and maintenance dosing period achieved or maintained sPGA (0,1)at rates of 68% (77% if excluding patients who had missing data owing to the COVID-19 pandemic) at week 60. This is similar to the rates of ≥69% reported in the pooled analysis of UNCOVER-1 and UNCOVER-2 and≥73% reported in UNCOVER-3.10Furthermore, in the present study, the majority of patients treated with ixekizumab in both treatment periods maintained or achieved near-complete skin clearance (PASI 90; 70% [79% when patients who had missing data due to the COVID-19 pandemic were excluded]) or complete clearance (PASI 100; 53% [60% when patients who had missing data due to the COVID-19 pandemic were excluded]) of psoriasis plaques through week 60. This is similar to the findings of UNCOVER-3, in which PASI 90 and PASI 100 response rates in ixekizumab-treated patients at week 60 were≥71% and ≥52%, respectively.10Such sustainable efficacy is important for effective clinical implementation of psoriasis treatment; results from a survey conducted in the United States indicate that rapid treatment response,maintaining skin clearance (for 2–3 years), and overall symptom relief are rated as highly important by >90% of psoriasis patients.7
Ixekizumab demonstrated rapid onset of action in Chinese patients receiving IXE Q4W and IXE Q2W compared with patients receiving placebo, as highlighted by improvements in PASI 75 at week 2. At week 4, around 60% of patients receiving ixekizumab achieved PASI 75.Rapid onset of action was also demonstrated by significant improvements in PASI 50 as early as week 1 and sPGA (0,1)as early as week 2. These improvements in disease severity were accompanied by rapid and significant improvements in Itch NRS by week 1 and DLQI by week 2.
The results of the present study show that the safety profile of ixekizumab in Chinese patients is consistent with that reported in the predominantly White patients included in the UNCOVER-1, UNCOVER-2,and UNCOVER-3 studies.1,10As expected, the majority of TEAEs were mild to moderate in severity. There were no deaths and few SAEs or discontinuations owing to TEAEs in patients treated with ixekizumab, neither in the safety population during the induction dosing period nor in the maintenance dosing period primary population during the maintenance dosing period. The most common TEAEs were upper respiratory tract infection and injection site reaction.
In clinical practice, comorbidities are key factors informing treatment selection for patients with psoriasis. Psoriatic arthritis is among the most common psoriasis-associated comorbidities. Ixekizumab received US Food and Drug Administration approval for the treatment of adults with active psoriatic arthritis in December 2017 and is recommended in this indication by Chinese and international treatment guidelines.13-15In addition, clinical studies have shown that ixekizumab can increase low-density lipoprotein levels and reduce high-sensitivity C-reactive protein levels without worsening aortic vascular inflammation or influencing glucose metabolism.16Furthermore, a pooled analysis of safety data from 7 studies of ixekizumab in psoriasis showed no increased risk of major adverse cardiovascular events.17The 2019 joint American Academy of Dermatology/National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis with biologics suggest that patients with psoriasis with a history of IBD or active IBD may experience disease recurrence or worsening with IL-17 inhibitors.18Therefore, although such adverse reactions are relatively rare in clinical trials, it is recommended that patients with psoriasis with a history of IBD or active IBD avoid IL-17 inhibitors.18Despite this, a pooled analysis of 4,209 patients with psoriasis who received ixekizumab across 7 clinical studies reported a <1% incidence of Crohn’s disease and IBD.17Finally, IL-17 inhibitors can be initiated in patients with chronic or resolved hepatitis under close monitoring of liver function tests andviral titers, and it is also safe to use IL-17 inhibitors in patients with latent tuberculosis infection.19

Table 3 AEs during the induction and maintenance dosing periods.
The maintenance dosing period of this study overlapped with the outbreak of the COVID-19 pandemic in China.No patients were infected with SARS-CoV-2 or diagnosed with COVID-19 during the study. The impact on patients was principally in the form of minor protocol deviations,including remote visits, visits outside the protocol window,a missed dose for 1 patient, and obtaining informed consent via telephone. Post hoc analysis revealed that missing efficacy measurements due to the COVID-19 pandemic did not change the efficacy conclusions of this study. The sPGA(0,1) and sPGA (0) response rates were higher in all intervention groupsvs. placebo when patients with missing data were excluded from the analyses at week 60. Although the COVID-19 pandemic disrupted the collection of laboratory tests and vital signs in some patients, this is not anticipated to have had a significant impact on the evaluation of the safety of ixekizumab.
The strengths of this study include the prospective, randomized, controlled, and multicenter study design and the high retention rate across all treatment groups, which minimized responder bias. A further strength was the use of physician- and patient-reported outcome measures,including assessments of health-related QoL. Utilizing physician- and patient-reported measures is clinically relevant given that many patients with psoriasis experience significant impairments in QoL.20One limitation of the present study is the relatively short duration given the chronic nature of psoriasis. This limitation will be mitigated by postmarketing surveillance that will allow for longer term monitoring of the efficacy and safety of ixekizumab in Chinese patients with moderate-to-severe psoriasis.
In conclusion, this study shows that both IXE Q4W and IXE Q2W result in superior efficacy compared with placebo after 12 weeks of treatment in Chinese patients with moderate-to-severe psoriasis. Importantly, clinical improvements in disease severity and patient QoL were rapid, and efficacy of ixekizumab was maintained for as long as 60 weeks. Safety findings in this patient population were consistent with the established safety profile of ixekizumab. Taken together, our findings support the use of ixekizumab in Chinese patients with moderate-to-severe psoriasis.
Source of funding
This study was sponsored by Eli Lilly, the manufacturer/licensee of ixekizumab. Eli Lilly was involved in the study design, data collection, data analysis, and preparation of the manuscript. Medical writing assistance was provided by Luke Carey, PhD, CMPP, and Koa Webster, PhD, of ProScribe–Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe’s services complied with the International Guidelines for Good Publication Practice.
Availability of data and material
Eli Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the United States and Europe and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents,including the study protocol, statistical analysis plan,clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.

- 國際皮膚性病學雜志的其它文章
- Characterization of Generalized Pustular Psoriasis in Northwest China: A Single-Center Retrospective Study
- Understanding the Pathogenesis of Generalized Pustular Psoriasis Based on Molecular Genetics and lmmunopathology
- Perspective on Melanoma in the Arab World:A Quantitative and Qualitative PubMed-Based Analysis of Research Output (2004–2019)
- Laboratory Safety of Dupilumab, and lts Effect on lnflammatory Biomarkers, in Chinese Adults With Moderate-to-Severe Atopic Dermatitis: An Analysis of a Randomized, Double-Blind Phase lll Study
- Sexual Behavior and Awareness of Sexually Transmitted Diseases Among Street-Based Female Sex Workers in the Florence Area, Central Italy
- Perceptions of Acne and Its Treatments Among Chinese College Students: A Cross-Sectional Survey