
C―C Coupling of Methane Mediated by Atomic Gold Cations under Multiple-Collision Conditions
2020-04-02 02:52YiRenQingYuLiuYanXiaZhaoQiYangShengGuiHe
物理化學學報 2020年1期
Yi Ren, Qing-Yu Liu, Yan-Xia Zhao, Qi Yang, Sheng-Gui He,*
1 State Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P.R.China.
2 University of Chinese Academy of Sciences, Beijing 100049, P.R.China.
3 Beijing National Laboratory for Molecular Sciences, Beijing 100190, P.R.China.
4 CAS Research/Education Center of Excellence in Molecular Sciences, Beijing 100190, P.R.China.
Abstract:The reactivity of atomic metal cations toward CH4 has been extensively investigated over the past decades.Closed-shell metal cations in electronically ground states are usually inert with CH4 under thermal collision conditions because of the extremely high stability of methane.With the elevation of collision energies, closed-shell atomic gold cations (Au+) have been reported to react with CH4 under single-collision conditions to produce andspecies.Further investigations found that the ion-source-generated cations can react with CH4 to synthesize C―C coupling products.These previous studies suggested that new products for the reaction of Au+ with CH4 can be identified under multiple-collision conditions with sufficient collision energies.However, the reported ion-molecule reactions involving methane were usually performed under single- or multiple-collision conditions with thermal collision energies.In this study, a new reactor composed of a drift tube and ion funnel is constructed and coupled with a homemade reflectron time-of-flight mass spectrometer.Laser-ablation-generated Au+ ions are injected into the reactor and drift 120 mm to react with methane seeded in the helium drift gas.The reaction products and unreacted Au+ ions are focused through the ion funnel and accumulate through a linear ion trap and are then detected by a mass spectrometer.In the reactor, the pressure is approximately 100 Pa, and the electric field between the drift tube and ion funnel can regulate the collision energies between ions and molecules.The reaction of the closed-shell atomic Au+ cation with CH4 is investigated, and the C―C coupling product is observed under multiple-collision conditions with elevated collision energies.Density functional theory calculations are performed to understand the mechanism of the coupling reaction Two pathways involving Au―CH2 and Au―CH3 species can separately mediate the C―C coupling process.The activation of the second C―H bond in each process requires additional energy to overcome the relatively high barrier (2.07 and 2.29 eV).Ion-trajectory simulations under multiple-collision conditions are then conducted to determine the collisional energy distribution in the reactor.These simulations confirmed that the electric fields between the drift tube and ion funnel could supply sufficient center-of-mass kinetic energies to facilitate the C―C coupling process to formThe following catalytic cycle could then be postulated: andThus, this study enriches the chemistry of both gold and methane.
Key Words:Gold; Methane;Ion-molecule reaction;C―C coupling;Mass spectrometry

1 Introduction
Methane is an appealing feedstock for production of valueadded chemicals because of plentiful resources of natural gas and flammable ice1-3.Carbon-carbon bond (C―C) coupling of CH4, which can synthesize higher hydrocarbons without intermediate steps, is an important paradigm for direct methane conversions4-6.Nevertheless, owing to the extremely high stability of methane with strong C―H bonds, negligible electron affinity, large ionization energy, and low polarizability, the direct conversion of CH4faces serious challenges.In gas-phase studies, the direct conversion of CH4 mediated by ionic species under isolated, controlled, and reproducible conditions is being actively studied7-13.
The reactivity of atomic transition-metal monocations (M+)towards CH4 has been extensively investigated, stimulated by wide application of transition-metal catalysts14-16.The groundstate M+ions are inert with CH4under thermal collision conditions, except for a few open-shell cations,i.e., Ti+, Nb+,Ta+, W+, Os+, Ir+, and Pt+, in which Ti+and Nb+react with CH4under multiple-collision conditions17-23.Thermodynamically and kinetically forbidden processes can become accessible by affording extra energies to reactants.Many atomic cations have been reported to become reactive toward CH4 under singlecollision conditions with high collision energies24-26.The closed-shell atomic Au+cations can react with CH4under singlecollision conditions to produceAuH+, andwith the elevation of collision energies in turn27.Further investigations by Schwarz and his co-workers have indicated that the ion source generated and thermalizedcations can react with CH4to synthesize C―C coupling products under single-collision conditions28.These studies imply that new products for the reaction of Au+with CH4can be identified under multiple-collision conditions with enough collision energies.In addition, it is essential to narrow the gap between gas-phase reactions and the “real-life” condense-phase reactions for direct conversion of CH4, which is usually operated under hightemperature (enough-collision energies) and high-pressure(multiple collisions) conditions29-31.The investigations of ionmolecule reactions under multiple-collision conditions with elevated collision energies are thus very important.
Traditionally, the ion-molecule reactions can be studied by flow tube reactor32-35, ion trap (IT) reactor36,37, Fourier-Transform ion cyclotron resonance (FT-ICR) reactor38, and guided ion beam (GIB) instrument39,40under either multiple-collision conditions with thermal collision energies or single-collision conditions with variable collision energies.A more complex condition, multiple-collision with variable collision energies,can be realized by utilizing a reactor composed of a drift tube(DT) and an ion funnel (IF).The DT, which has been extensively used as the analyzer in the ion mobility spectrometry41,42, as well as the reactor of proton transfer reactions43,44and electron attachment reactions45,46have been used to study the interactions of Ti+, Co+, and Er+with CH423,47,48.In the DT, ions drift under an adjustable direct current (DC) electric field accompanying multiple collisions with neutral molecules.The IF is usually used to couple with the DT for efficient capture and focus of ions to pass through a conductance limiting orifice49-54.
In this work, a reactor composed of a DT and an IF has been constructed and attached to a reflectron time-of-flight mass spectrometer (TOF-MS) equipped with a laser ablation ion source and an ion trap.By using the apparatus, at the pressure around 100 Pa, the reactions of Au+with CH4under multiplecollision conditions with variable collision energies have been studied and the reaction channels for C―C coupling products have been identified, enriching the chemistry of Au+and CH4.
2 Experimental and theoretical methods
2.1 Experimental methods
The reaction of Au+with CH4was performed by a homemade reflectron TOF-MS equipped with a laser ablation ion source, a DT-IF reactor and an ion trap in this study.The schematic diagram of the apparatus is shown in Fig.1.The laser ablation ion source (Part 1) has been described in our previous works55,whereas the DT-IF reactor (Parts 2 and 3), the ion trap (Part 7),and the reflectron TOF-MS (Parts 9-13) were newly designed in this work.The details of the experimental apparatus are given below.
2.1.1 Laser ablation ion source
The laser ablation ion source, which was used to generate atomic Au+cations, was attached to the DT-IF reactor by a tube adaptor.Briefly, a 532 nm [second harmonic of a Nd3+: yttrium aluminum garnet (Nd : YAG)] laser (green line in Fig.1) with an energy of 10 mJ/pulse and a repetition rate of 10 Hz was focused on a rotating and translating gold disk.In the presence of pure helium carrier gas (99.999%), atomic Au+cations were generated and expanded into the DT-IF reactor against an electric field.The carrier gas of He was controlled by a pulsed valve(General Valve, Series 9) with a backing pressure of about 4 ×105Pa and formed an apparent pressure of about 10 Pa in the DT-IF reactor.The number of collisions between Au+and He before the arrival of Au+at the reactor is calculated by hardsphere model.With a collision cross section of 0.09 nm2and a helium pressure of about 5000 Pa in the ion source, an Au+cation will experience a collision rate of 4 × 108s-1with helium,resulting 104collisions in about 25 μs.The Au+could be well thermalized to the ground electronic state (5d10,1S0).It is noteworthy that Armentrout and his coworkers27,56reported that the electronically excited states of Au+(6s15d9,3D1-3 and1D2)had been observed to survive ~105thermalizing collisions with He and then ~104collisions with Ar.The GIB experiment indicated that at thermal collision energies, the ground state Au+was very un-reactive with methane (CD4was used in the experiment)27while the excited state Au+could readily react with CD4 to form product ions AuD+, AuCD+2, and(relatively weak).The Au+ions in the previous studies27,56were generated by a direct current discharge flow tube (dc/FT) source while the Au+ions in this study were generated by the laser ablation.To confirm that the Au+ions generated from our laser ablation source were in the ground electronic state, the Au+ions from the source were directly collected and confined (additional~1000 collisions with He)57in the ion trap to react with a pulse of CH4gas.It turned out that none of AuH+,orcould be observed and only the association product ionsandcould be generated (see the Results and Discussion section below).One may conclude that the Au+ions from the laser ablation source are much cooler than those from the dc/FT source.

Fig.1 A schematic diagram of the reflectron time-of-flight mass spectrometer coupled with a laser ablation ion source, a reactor composed of a drift tube and an ion funnel, and a linear ion trap.
2.1.2 DT-IF reactor
The DT-IF reactor was used to realize the reactions between Au+and CH4under multiple-collision conditions with variable collision energies.The DT (Part 2) consisted of 12 uniform 1.0 mm thick stainless steel ring electrodes with an outer diameter of 80.0 mm and an inner diameter of 25.0 mm.These electrodes were coaxially arranged by four stainless steel screws (Part 17)covered with polyetheretherketone (PEEK) tubes (Part 19) and uniformly spaced by 9.0 mm thick PEEK spacers (Part 18).All electrodes in the DT were consecutively connected by 1.0 MΩ metal film resistors.The potentials of head and rear electrodes were independently controlled by two DC power supplies (0-200 V), which caused a uniform electric field inside DT.
The IF (Part 3) consisted of 47 0.5 mm thick stainless steel electrodes.The inner diameters of these electrodes were decreased linearly from 25.0 mm to 2.0 mm.To reduce the total capacitance, the excess material on each electrode had been trimmed.The electrodes were separated by 0.5 mm thick polytetrafluoroethylene (PTFE) washers (Part 20) producing a gastight arrangement (apart from the 2.0 mm aperture).Tabs of electrodes were used for electrical connection with a custommade circuit board consisting of 48 1.0 MΩ metal film resistors and 47 120 pF capacitorsviatwo zero insertion force (ZIF)connectors.The resistors ensured a same DC voltage gradient between neighboring electrodes, and capacitors were utilized to decouple the DC and radio frequency (RF) voltages58.The RF voltages of equal amplitude but opposite phase applied to the IF adjacent electrodes were 20-160 Vp-p (peak to peak voltage)with frequency of 1.17 MHz and delivered through a handmade air-core transformer which was driven by a commercially available RF solid state amplifier (Amplifier Research,75A250A) connected with a waveform generator (RIGOL,DG1022U).The potential of head electrode was independently controlled by utilizing a DC power supply from 2 to 60 V, and the potential of rear electrode was constant, 2 V referenced to ground.
The stacked ring RF ion guide (Part 4), a transition between the reactor and a skimmer (Part 5), consisted of 5 uniform 0.5 mm thick electrodes with a 6 mm center bore.The stack of electrodes was performed by using 2.5 mm thick PEEK spacers(Part 21) for pumping the gas from the reactor.The RF generation system of the stacked ring RF ion guide was the same as the one applied to IF.The RF voltages of equal amplitude but opposite phase are applied to the adjacent electrodes.The amplitude of RF applied to the stacked ring RF ion guide was about two times of the one applied to the IF.The DC bias of each electrode was 2 V referenced to ground.
The DT, IF, and stacked ring RF ion guide were assembled to form a DT-IF reactor.The distance between the rear electrode of DT and the head electrode of IF was 10 mm.The DC potential difference between the rear electrode of DT and the head electrode of IF can be independently varied to accelerate(activate) the ions from DT to react with CH4downstream.Similar configuration of applying the DC voltages was used to study conformer transitions of biologically relevant species by ion mobility mass spectrometry53,54.The drift/reactant gas,0.1%-10% CH4seeded in He (99.999%), was delivered into the DT-IF reactorviaa 4 mm inner diameter stainless steel tube (Part 14) which had been soldered to DC cell chamber near the ion source.The pressure of drift/reactant gas in the reactor was monitored by a capacitance manometer and maintained around 100 Pa by a metering valve.The drift/reactant gas was finally extracted by the pump of Chamber I in which the pressure was estimated to be 10-1Pa.
2.1.3 Ion trap
The ion trap (Part 7) was used to accumulate the ions released from the reactor.The structure of ion trap (Fig.2a) was described in our previous works58, but the trap was run at a different mode with respect to the previous one58.Three sets of pulsed DC potentials shown asU1,U2, andU3in Fig.2c-e were applied to two cap electrods (1, 2 in Fig.2a) and hexapole rods (3 in Fig.2a), respectively (note that U3includes the superimposed RF components with amplitude about 250 Vp-pat 770 kHz).Before the reactant and product ions arrived at the trap, a pulse of cooling gas of He (99.999%) had been delivered into the cell through a pulsed valve.TheU1andU2were different positive DC potentials (typically 3 V and 30 V, respectively), and the average ofU3was 0 V referenced to ground.The ions with enough kinetic energy against the potential of the front cap electrode, could enter into the trap, and then were cooled by collisions with He gas and confined in the trap.After accumulating the ions for about 5 ms (further longer time did not increase the ion signal significantly), theU1andU3were elevated for preparing the ejection of the ions.AsU2was decreased to 0 V, the ions were ejected from the trap and focused into the reflectron TOF-MS.Then,U1,U2, andU3 were restored for the next period.More ions could be delivered into the reflectron TOF-MS by the accumulating mode of the ion trap,which improved the sensitivity of the apparatus by about one order of magnitude compared with the normal mode to run the trap in our previous study, whereU1was set to 0 V for a short period (< 100 μs) of time to collect the ions58.
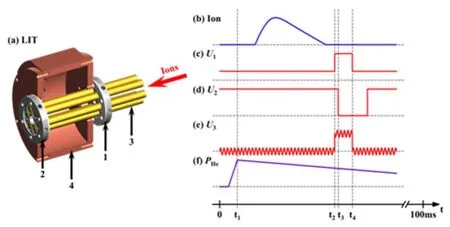
Fig.2 A schematic diagram of a linear ion trap (a) and the pulsed events (b-f) to run the trap.
2.1.4 Reflectron TOF-MS
The reflectron TOF-MS was used to detect the reactant Au+and the product ions of the reaction.For enhanced space efficiency, all of the parts of TOF-MS, including the electrodes for accelerating ions (Part 9 in Fig.1), the deflectors (Part 10),the Einzel lens (Part 11), the reflector (Part 12), and the dual microchannel plate (MCP) detector (Part 13), were coaxially arranged.The ions (red line in Fig.1) passed through Part 9 (the right two pieces of the three electrodes), 10, and 11 twice.Pulsed potentials were applied to Part 9 and Part 10.The ions could be focused by the Einzel lens twice under a constant potential.The signals from the dual MCP detector were recorded by a digital oscilloscope with a mass resolution (m/Δm) of about 1300 for Au+.
2.2 Theoretical methods
2.2.1 Ion trajectory simulations
The SIMION 8.1 software59were used to study the ion trajectories of Au+in the DT-IF reactor with 100 Pa He (note that 0.5%-5% CH4in He was used in the experiment).The hardsphere collision model was adopted to treat each collision event between Au+and He.The experimental parameters of the DC and RF electric fields were used.The voltage between DT and IF (UDT-IF) was varied and 2000 trajectories of Au+were calculated for each experimental condition.The speed of Au+in 100 Pa He was recorded and the center-of-mass kinetic energy between Au+and CH4was calculated and analyzed.
2.2.2 Potential energy surface calculations
Density functional theory (DFT) calculations with the Gaussian 09 program60were performed to study the C―C coupling of two CH4 molecules mediated with the Au+ion.The TPSS (Tao-Perdew-Staroverov-Scuseria) functional61with good performance for bond energies62,63of Au+―C, Au―H, C―H,C―C, and H―H, as well as reaction barriers of several systems64were chosen in this work.The 6-311+G(d) basis sets65for C, and H atoms and D95V basis sets66connected with the Stuttgart/Dresden relativistic effective core potentials (denoted as SDD in Gaussian software) for Au atom were used in all of the calculations.The B3LYP calculations with the same basis set were also performed for selected species to compare with the TPSS method.The reaction mechanism calculations involved the geometry optimization of reaction intermediates (IMs) and transition states (TSs) through which the IMs transfer to each other.The TSs were optimized using the Berny algorithm method67, and the initial guess structures of the TSs were generally obtained through relaxed potential energy surface(PES) scan using appropriate internal coordinates68.The vibrational frequency calculations were executed to check that the reaction IMs and TSs have zero and only one imaginary frequency, respectively.Intrinsic reaction coordinate (IRC)69,70calculations were performed to verify that the TS connects two appropriate local minima in the reaction.The zero-point vibration corrected energies (ΔH0) are reported in this work.
3 Results and discussion

Fig.3 TOF mass spectra for the reactions of Au+ with (a) He,(b) 0.5% CH4 seeded in He, (c) 5.0% CH4 seeded in He, and (d) 2.0%CD4 seeded in He in the DT-IF reactor at the drift pressure about 100 Pa.TOF mass spectrum for the reaction of Au+ with CH4 in the ion trap reactor was shown in (e).
The typical TOF mass spectra for the interactions of laser ablation generated Au+cations with CH4 and CD4 are shown in Fig.3.The experimental conditions for the DT-IF reactor were:(1) 4.2 V·cm-1DC voltage gradient of DT (GradDT), (2) 60 V voltage between DT and IF (UDT-IF, 60.0 V·cm-1DC voltage gradient), (3) 1.0 V·cm-1DC voltage gradient of IF (GradIF), and(4) 99 Vp-pRF amplitude of IF (AmplIF).Upon interactions of Au+with CH4 (0.5% in 100 Pa He for Fig.3b and 5.0% in 100 Pa He for Fig.3c) in the DT-IF reactor, products assigned as(very weak),andwere observed.In addition, the weak association productsand (AuC2H4)H2O+were also produced (not marked in Fig.3c).The water, which adsorbed on the ions,originated from trace amount of impurity in the gas-handling system.The isotopic labelling experiment with CD4 (Fig.3d)further confirmed the assignments of the products in Fig.3b,c.It should be noted that if the ion source generated Au+was directly injected into and confined in the ion trap to interact with CH4,only the association productscould be observed and product ions such as AuH+anddue to collision induce dissociation or excited state (Au 6s15d9)reaction were completely absent (Fig.3e).
The primary reactions in the DT-IF reactor are listed below:

Formation of AuH+andis due to reaction of Au+with CH4at high collisional energies (see the result from ion trajectory simulations below) because Au+cannot cleave the C―H bond of CH4 under thermal collision conditions (Fig.3e)while AuH+andwere observed in the GIB experiment at center-of-mass collisional energies above 2 eV27.In addition to AuH+andthe GIB experiment on Au+/CH4 also identifiedat even lower center-of-mass collisional energies (~1.0 eV).However,was relatively very weak with 0.5% CH4in the reactor (Fig.3b).Note that very weakcould also be observed in the 2% CD4experiment (Fig.3d).A reasonable explanation is that althoughions were generated in our experiment, they rapidly reacted with CH4to form the C―C coupling product(reaction 2, the rate constant ofcm3·molecule-1·s-1)according to the investigations of Schwarz and co-workers28.The reaction of theintermediate with CH4(reaction 2) is also supported by the result that the ion signal ofalmost disappeared completely at the high methane concentration (5%in He, Fig.3c).


The TOF mass spectra for the reactions of Au+with CH4under differentUDT-IFvoltages are shown in Fig.4.At lowUDT-IF, the association specieswere the main products due to enough cooling by collisions with 100 Pa He(collision rate is about 107s-1).As the increase ofUDT-IF, the relative ion intensities ofdecreased while the relative ion intensity ofincreased, which suggests thatcan either dissociate back toor transfer directly tothrough reaction 3.The high value ofUDT-IF accelerates both Au+andions.The accelerated Au+can result in reaction 1e and then reaction 2 that generatein the IF.The acceleratedmay also generatethrough reaction 3.
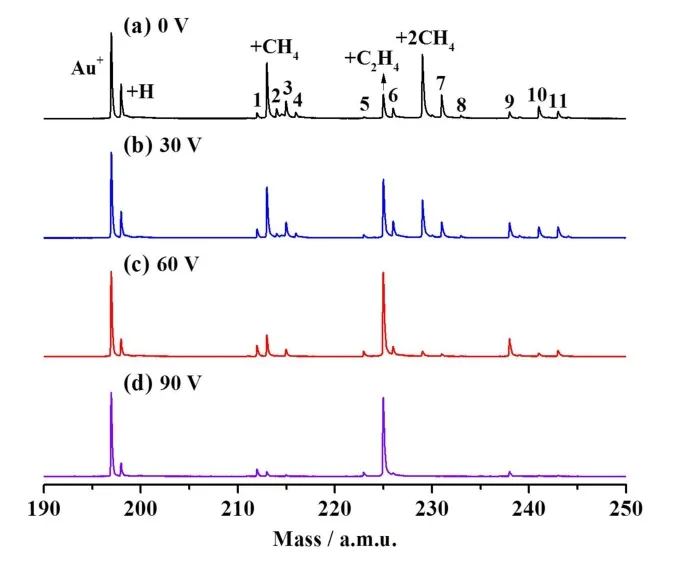
Fig.4 TOF mass spectra for the reactions of Au+ with 5.0% CH4 seeded in 100 Pa He under UDT-IF values of 0 V (a),30 V (b), 60 V (c), and 90 V (d).
The ion trajectory simulations were used to analyze the centerof-mass collisional energies (Ec) provided to the reaction system at differentUDT-IF values.The distribution ofEc values between Au+and CH4with respect to theUDT-IFvalues and the ion positions along the axis of the DT-IF system is shown in Fig.5.AtUDT-IF= 0 V (Fig.5a), theEcvalues are all small (typically less than 0.5 eV) in the reactor except at the exit region of the IF.Some of the Au+ions at the IF exit have highEc values up to 3-4 eV, which can lead to formation of AuH+(reaction 1c) and(reaction 1d) even at theUDT-IFvalue of 0 V (Fig.4a).The relative intensity of AuH+is higher than that ofwhich is consistent with the GIB experiment.27When theUDT-IFvalue increases from 0 V to 30 V (Fig.5b), theEcvalues for the ions in between the rear electrode of the DT and the head electrode of the IF (the region between the two dashed lines in Fig.5, such region is denoted asacceleration regionin the text below) increase significantly and some of theEcvalues are as high as 1.5 eV.The averageEcvalue () for the ions in the acceleration region was analyzed and shown in Fig.6.There is a good linear-dependence betweenandUDT-IF.The average number of collisions between Au+and CH4 (5% CH4 in 100 Pa He) in the acceleration region is around 5, so multiple-collisions with relatively highEcvalues (1.5-2.0 eV forUDT-IF= 60 V, see Fig.5c) could take place in the experiment to result in processes such as reaction 3.

Fig.5 Distribution of center-of-mass collisional energies (Ec)between Au+ and CH4 with respect to the relative positions of the ions along the axis of the DF-IF reactor at the UDT-IF values of 0 V (a),30 V (b), 60 V (c), and 90 V (d).
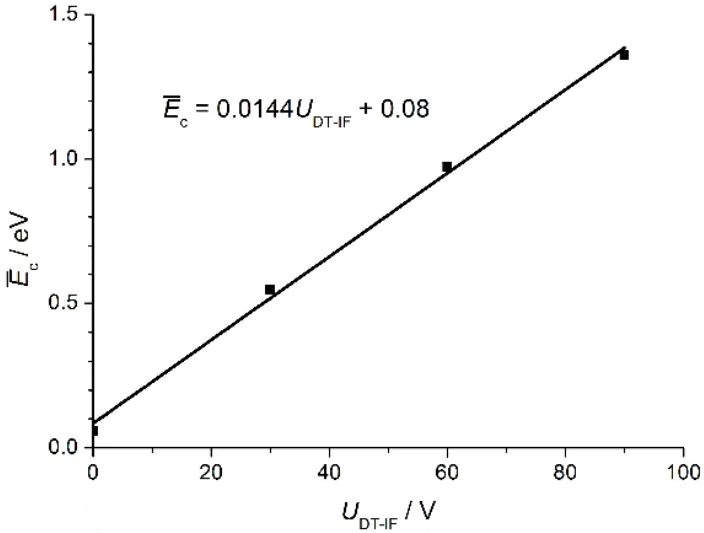
Fig.6 The average collisional energy () for the ions in the region denoted by the dashed lines in Fig.5.
The mechanisms of reactions 1e and 2 that can lead to formation ofhave been separately investigated by the research groups of Armentrout and Schwarz, respectively27,28.The mechanism of reaction 3 has been studied by the DFT calculations in this work and the result is shown in Fig.7.Fig.7a shows the reaction pathway involving Au―CH2 species.The Au+ion traps two CH4to form encounter complex 2 (R→1→2).Two H atoms of CH4in 2 can transfer to the Au atom and form H2molecule in 3 (2→3).This process is similar to the one for formingin Ref.27.Then, the C―C coupling takes place(3→3/4→4) and C2H6molecule is formed in 4.In the intermediate 4, two H atoms have a distance of 84 pm which is close to the H-H bond length (74 pm) in the free hydrogen molecule, suggesting that H2 formation is nearly complete and H2molecule can be evaporated from 4 (4→5).The intermediate 5 can be considered as the encounter complex of Au+ion with C2H6molecule.The dehydrogenation of C2H6by Au+producesand H2, which has been described previously28,71.The intermediate 3 can also transform into 8 by desorption of the H2unit (3→8).The intermediate 8 needs extra energy (0.52 eV) to transfer CH4moiety from Au atom to CH2moiety (8→8/9→9).The coupling of CH4and CH2then takes place (9→9/5→5),which has been well described in Ref.28.The relative energies ofwith respect to Au++ CH4are also shown in Fig.7a.The ion trajectory simulations indicate that theEcvalues can be as high as 1.5 eV atUDT-IF = 60 V (Fig.5c).Note that the highest barrier in Fig.7a is 2.17 eV [ΔH(3/4) - ΔH(2)].Such a barrier can be well overcome by the total energy ofEc(1.5 eV) and the binding energy (1.08 eV) between 1 and CH4.This theoretical result is consistent with the experimental observation of significant signal increase of AuC2H4+atUDT-IF= 60 V with respect toUDT-IF= 0 eV in Fig.4.

Fig.7 TPSS calculated potential energy profiles for the reaction of Au+ with two CH4 to generate AuC2H4+ and two H2 molecules involving Au―CH2 (a) and Au―CH3 (b) as the intermediates.
The previous studies of Armentrout, Schwarz, and their coworkers indicated that for the formation ofthrough reactions 1e and 2, onceis generated the C―C coupling process (reaction 2) is facile28.The GIB experiment indicated that the reactionis endothermic by(0.96 ± 0.05) eV that is very close to the value of 0.97 eV by the B3LYP calculations in which the Au+ion simultaneously activates two C―H bonds of the CH4 molecule.Note that the TPSS method underestimated the endothermicity ofby 0.36 eV (1' in Fig.7a)27.The mechanism and energetics of CH4activation by Au+are generally similar to the result shown in Fig.7a for reaction 3: (1) thecomplex simultaneously activates two C―H bonds of the second CH4molecule and (2) the reactionis endothermic by 0.99 eV at the TPSS level[the value is 1.33 eV by B3LYP calculations, note that the desorption of H2from Au(CH4)(CH2)(H2)+is facile].As a result,the pathway shown in Fig.7a for reaction 3 was predicted to be slightly less favourable by 0.3-0.4 eV for the activation of the first CH4 molecule than the reaction couple (1e and 2) to produce
The reaction pathway for reaction 3 involving Au―CH3species is shown in Fig.7b.The reactions from the encounter complexes 1 and 2 can proceed through the mechanism of oxidative addition (1→1/10→10 and 2→2/11→11), resulting in the activation of the first C―H bond and formation of the Au―CH3and Au―H bonds.Note that 10 can trap CH4to form 11.The intermediate 11 needs extra energy (0.66 eV) to activate another C―H bond in the second CH4 (11→11/12→12) to form the second Au―CH3bond and H2formation is nearly complete in 12 (the distance of two H atoms is 77 pm).After the evaporation of H2molecule from 12 (12→13), the C―C coupling takes place (13→13/5→5) and C2H6 molecule is formed in 5 that can finally transform into(Fig.7a).The energy of the transition state 11/12 (0.66 eV) is higher than that of the transition state 3/4 (-0.05 eV), so the pathway of CH2/CH4coupling (Fig.7a) is more favourable than the pathway of CH3/CH3coupling (Fig.7b).
The DFT calculations (Fig.7) indicate that theintermediates such 5, 6, and 13 are formed to produceHowever, the product ionwas not observed in the experiments (Figs.3 and 4).In the case that theUDT-IFvalue (and theEcvalue, see Fig.6) is high enough to overcome the barriers of 1→3/4 (1.09 eV) and 1→11/12 (1.80 eV), the formedcan be subsequently activated directly to lose H2 so thatis formed or thespecies such as 5 can react with CH4to formnote thatUDT-IFsupplies extra energies].
The DFT calculations indicated that direct dissociation of AuC2H4+into C2H4and Au+needs extra energies of 2.98 eV.It is possible that the substitution reactionmay take place with extra energies in the experiment, then a catalytic cycle can be complete:

The above cycle is very similar to the one that have been well studied by Bernhardt, Landman, and their co-workers for the same reaction catalyzed by thespecies72.Note thatand Au+have open-shell and closed-shell electronic structures,respectively.As reported in Ref.72, thesystem mediates the C―C coupling through coupling of CH3and CH2species.In contrast, the Au+system can mediate the same process through coupling of CH2with CH4(reaction 2 and the reaction path of Fig.7a) or coupling of two CH3species (reaction path of Fig.7b) when the reaction system has enough extra energies.
4 Conclusions
A reactor composed of a drift tube and an ion funnel has been built to couple with a laser ablation ion source, an ion trap, and a reflectron time-of-flight mass spectrometer.The apparatus has been used to study the reaction of the Au+ions with CH4under multiple-collision conditions with variable collision energies.The association productscan be formed in the reactor with relatively high gas pressure (100 Pa).The electric fields between the drift tube and the ion funnel can supply enough center-of-mass kinetic energies to facilitate the C―C coupling process to formThis study is among the first to report the C―C coupling of two CH4molecules mediated with a closed-shell atomic cation (Au+).The quantum chemistry calculations have indicated that the C―C coupling process in reactioncan involve both Au―CH2and Au―CH3species as the intermediates.The collisional energy distributions provided by ion trajectory simulations further support the experimental results.Though the experimental conditions are still far from the “real-life”condense-phase reactions, this study is a small step to narrow the gap between gas-phase and condense-phase reactions and enriches the chemistry of both gold and methane.
