
Function and dysfunction of GEMIN5: understanding a novel neurodevelopmental disorder
2024-03-05 08:49CharlesNelsonUdaiPandey
中國神經再生研究(英文版) 2024年11期
Charles H.Nelson ,Udai B.Pandey,2,*
Abstract The recent identification of a neurodevelopmental disorder with cerebellar atrophy and motor dysfunction (NEDCΑM) has resulted in an increased interest in GEMIN5,a multifunction RNA-binding protein.As the largest member of the survival motor neuron complex,GEMIN5 plays a key role in the biogenesis of small nuclear ribonucleoproteins while also exhibiting translational regulatory functions as an independent protein.Although many questions remain regarding both the pathogenesis and pathophysiology of this new disorder,considerable progress has been made in the brief time since its discovery.In this review,we examine GEMIN5 within the context of NEDCΑM,focusing on the structure,function,and expression of the protein specifically in regard to the disorder itself.Additionally,we explore the current animal models of NEDCAM,as well as potential molecular pathways for treatment and future directions of study.This review provides a comprehensive overview of recent advances in our understanding of this unique member of the survival motor neuron complex.
Key Words: cerebellar atrophy;GEMIN5;neurodevelopmental disorder;neurological disease;SMN complex
Introduction
The survival motor neuron (SMN) complex is a multi-protein construct vital to the ATP-dependent biogenesis of small nuclear ribonucleoproteins (snRNPs) (Meister et al.,2001).The SMN complex recruits snRNP precursors,specifically small nuclear RNAs (snRNAs),and transfers a ring of Smith(Sm) proteins,often referred to as the Sm core,to the snRNA molecule (Meister et al.,2001;Neuenkirchen et al.,2015).While the SMN complex is responsible for ensuring the correct binding of the Sm core at the Sm site,a short,highly conserved nucleotide motif found within snRNA,it relies on the protein arginine N-methyltransferase 5 (PRMT5)complex for Sm core construction (Neuenkirchen et al.,2015).Following the creation of this snRNA-Sm core construct,a nuclear localization signal is added by the SMN complex,allowing both the construct and the SMN complex nuclear entry whereupon they will separate and localize to specific nuclear bodies (Mouaikel et al.,2003).Finally,the snRNΑSm protein construct is further modified to become a mature snRNP where it will then be integrated into the spliceosome via a stepwise construction process before ultimately playing a major role in mRNΑ processing (G?rnemann et al.,2005).The human SMN complex comprises nine distinct components:SMN,GEMINs 2–8,and UNRIP (Otter et al.,2007).WhileSMN1mutations are notable for their involvement in spinal muscular atrophy (SMA),an autosomal recessive disease that affects muscular control and the peripheral nervous system(Lefebvre et al.,1995),the pathology associated withGEMINmutations has only recently begun to receive significant attention.
The GEMINs,fully named as the gem nuclear organelle associated proteins due to their association with nuclear gems (Gemini of Cajal bodies),are a functionally diverse group of proteins most consistently observed as components of the SMN complex (Liu and Dreyfuss,1996;Meister et al.,2001;Gubitz et al.,2002).To date,seven GEMIN proteins are known and numbered GEMIN2 to GEMIN8 with SMN as the de facto “GEMIN1.”In recent years,rare pathogenic variants of GEMIN5 have been linked to a novel neurodevelopmental disorder currently referred to as Neurodevelopmental Disorder with Cerebellar Atrophy and Motor Dysfunction(NEDCΑM) (OMIM # 619333) or simply as GEMIN5 syndrome.While only first reported in 2021,a total of 48 cases have been published in the literature at time of writing with each case possessing pathogenic,rare genetic variants ofGEMIN5in a compound heterozygous or homozygous manner (Kour et al.,2021;Saida et al.,2021;Francisco-Velilla et al.,2022b;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).
While the chaperone function of the SMN complex is impaired by mutant SMN or GEMIN5 proteins,the distinctive disease phenotypes suggest other functions for one or both of these proteins outside of those related to the SMN complex (Kour et al.,2021;Saida et al.,2021;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).This is significant in that it not only increases understanding of the SMN complex’s chaperone function,but also furthers understanding of human neurodevelopmental processes in general.Thus,as a new branch of research has begun into other proteins within the SMN complex,this review aims to comprehensively examine GEMIN5 within the context of NEDCΑM,addressing structural,functional,and expressional aspects as well as current animal models,potential treatment pathways,and recent advancements in our comprehension of this distinct SMN complex constituent.
Search Strategy
In searching for relevant literature,PubMed,Google Scholar,and the University of Pittsburgh’s Health Sciences Library System were most used.Searching began in late 2022 and continued through the drafting period.Search modifiers such as “cerebellum,”“SMA,”“SMN,”“mice,”“Drosophila,”and “zebrafish”were used.As the primary objective of this narrative review is to comprehensively examineGEMIN5,especially within the context of NEDCAM,articles providing key information about the discovery,structure,function,and dysfunction ofGEMIN5and its various orthologs were selected.Additionally,works that provided key insights,investigated NEDCAM,or provided important contextual information about the topic were also selected.
GEMIN5 Structure,Localization,and Expression
Found at human chromosomal location 5q33.2,GEMIN5is a well conserved,universally expressed gene comprising 28 exons and encoding a canonically 1508 amino acid long RNΑ binding protein (RBP) (Gubitz et al.,2002;Martinez-Salas et al.,2020).As with mostGEMINs,GEMIN5was initially investigated due to its relationship withSMNand was first identified in 2002 by co-immunoprecipitation techniques with other members of the SMN complex (Gubitz et al.,2002).The expressed GEMIN5 protein possesses three distinct domains(Figure 1).The first domain comprises 13 tryptophan-aspartic acid (WD-40) repeats that form two seven-bladed propellerlike structures on the amino terminus (Gubitz et al.,2002;Jin et al.,2016).These structures facilitate pre-snRNA binding with the Sm site and m7G cap of the RNΑ,a chaperone activity vital to correct SMN complex function (Lau et al.,2009;Xu et al.,2016).The second domain possessed by GEMIN5 is a tetratricopeptide-like (TPR-like) domain comprising 17 alpha-helices in a coiled-coil motif,allowing for self-assembly and the creation of GEMIN5 dimers (Moreno-Morcillo et al.,2020).The domain is named for its similarity to the tetratricopeptide repeat domain and was first identified within elongator complex protein 1 (ELP1),where it also functions as a dimerization domain (Xu et al.,2015).Interestingly,unlike the TPR-like domain found in ELP1 which dimerizes as a horseshoe-like shape,the GEMIN5 TPR-like domain differs in conformation and creates a unique canoe-like structure upon dimerization (Xu et al.,2015;Moreno-Morcillo et al.,2020).Although evidence suggests that disruption of the domain hampers ribosomal interactions,it is not known to facilitate any additional protein-protein or protein-RNA interactions(Moreno-Morcillo et al.,2020).Regardless,the domain’s high degree of conservation between GEMIN5 orthologs suggests a pivotal role in maintaining both structure and function (Xu et al.,2015;Martinez-Salas et al.,2020;Moreno-Morcillo et al.,2020;Francisco-Velilla et al.,2022b).

Figure 1|A schematic of GEMIN5 protein illustrating the three different functional domains with pertinent structural and functional information.
The final domains present in GEMIN5 are the non-canonical RNA binding sites (RBSs),designated RBS1 (residues 1297–1412) and RBS2 (residues 1383–1508) (Fernandez-Chamorro et al.,2014).While both RBSs are referred to as non-canonical due to their differences when compared to other known RNΑ binding motifs,each site differs in RNA binding capacity as well as binding targets (Fernandez-Chamorro et al.,2014;Francisco-Velilla et al.,2018).In addition,the RBS1 structural domain appears to be intrinsically unstructured,indicating that the domian lacks secondary structure with or without bound RNA,whereas the RBS2 structural domain has several predicted helices,though this remains to be confirmed experimentally (Francisco-Velilla et al.,2018;Embarc-Buh et al.,2021).While the molecular specificities of the RBS2 site are not yet well understood,research into the RBS1 site has shown that it interacts with the stem loop regions of both viral and cellular RNΑ via highly conserved arginine and aromatic residues (Martinez-Salas et al.,2020;Embarc-Buh et al.,2021).These residues then facilitate the domain’s ability to engage in selective translational regulation,a process which appears to be highly dependent on the secondary structure of the RNA transcript (Francisco-Velilla et al.,2018;Martinez-Salas et al.,2020).RBS1 in particular has shown a high affinity forGEMIN5mRNA,allowing for an elevated degree of autoregulation with regards to protein expression (Francisco-Velilla et al.,2018).Surprisingly,recent evidence has shown that loss of the RBSs significantly perturbs the ability of GEMIN5 to bind and interact with SMN,a process that occurs via interaction with the latter protein’s Tudor domain (Fortuna et al.,2023).This suggests that the RBSs also facilitate protein-protein interaction,though the mechanism is not yet well defined.Αlso of note,GEMIN5 possesses an RKΑR motif,making the protein susceptible to proteolysis by picornavirus L protease(Pi?eiro et al.,2012).While this motif is shared by a number of viral host factors,the motif is relevant as proteolysis results in the creation of two stable,though not fully defined fragments: p57 and p85 (also referred to as G5C) (Pi?eiro et al.,2012;Guo et al.,2022).The G5C fragment,thought to comprise the TPR-like and RBS domains,has been of recent interest as it is able to bind with both full-length GEMIN5 as well as pentamerize with other G5C fragments (Moreno-Morcillo et al.,2020;Guo et al.,2022).This pentamer solely contains alpha helices and can then further dimerize with itself,creating a homodecamer (Guo et al.,2022).It appears that this conformation allows the fragment to bind to the stem loop regions of certain RNA targets,however,the full significance of this stable fragment and homodecamer are not yet fully understood,especially given the importance of both domains to GEMIN5 function.
Unlike other members of the SMN complex,GEMIN5 is a primarily cytoplasmic protein with some additional nuclear presence (Figure 2CandD),however the protein has also been observed to be concentrated within specific nuclear and cytoplasmic bodies (Gubitz et al.,2002;Hao et al.,2007;Cauchi et al.,2010).Localization within nuclear gems is observed,however unlike most SMN complex members,colocalization between SMN and GEMIN5 is scarce,implying that GEMIN5 is no longer necessary to SMN complex function when in the nucleus (Battle et al.,2007;Hao et al.,2007).Within the cytoplasm,GEMIN5 can be found in association with the SMN complex during Sm core ring construction.Alternatively,the protein can be found associating with GEMIN3,GEMIN4,or completely independent (Battle et al.,2007;Hao et al.,2007).GEMIN5 ortholog localization has also been observed withinDrosophilacytoplasmic U-bodies,most likely due to its role in U-rich snRNP biogenesis as their presence is the defining feature of such bodies (Liu and Gall,2007;Cauchi et al.,2010).Further,GEMIN5 has also been shown to associate with and potentially influence the formation of P-bodies,an interesting though not entirely unexpected observation given the connection with U-bodies(Fierro-Monti et al.,2006;Xu et al.,2016;Jiang et al.,2018;Martinez-Salas et al.,2020).The protein is also recruited into both RNP-and CAPRIN1-associated cytoplasmic granules as part of stress response,though the significance of this localization is unknown (Battle et al.,2007;Vu et al.,2021;Delle Vedove et al.,2022).
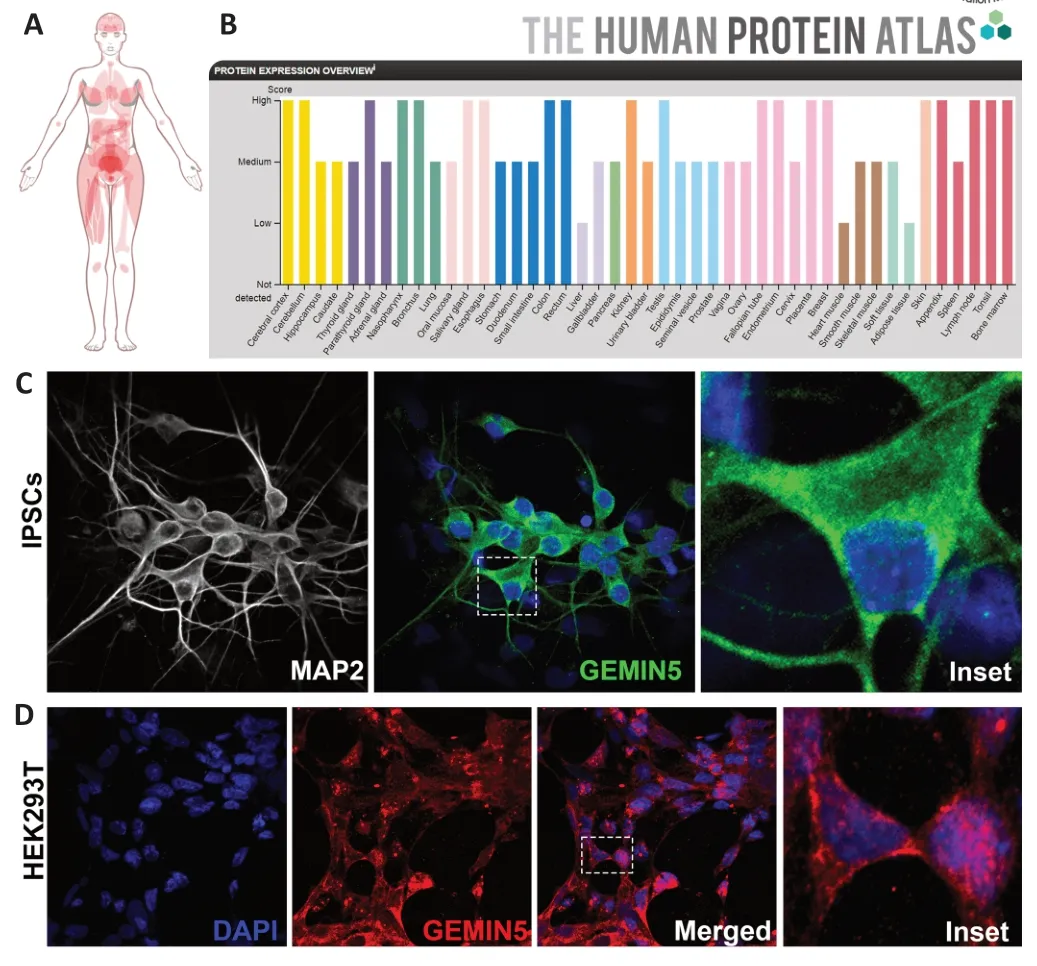
Figure 2| Expression and localization of endogenous wild-type GEMIN5 protein within humans.
Regarding expression between tissue types,GEMIN5 is a ubiquitously expressed protein with little to no tissue specificity,though some exceptions are present.Based on data from The Human Protein Αtlas (Uhlén et al.,2015),GEMIN5 protein is characterized as “low”within the liver,heart muscle,and adipose tissue,with “medium”to “high”expression in all other listed tissue types.Notably,GEMIN5 expression appears to be high in the human cerebral cortex and the cerebellum,especially within Purkinje cells(Uhlén et al.,2015).While further studies may increase our understanding of GEMIN5 expression patterns in varioustissue types,expression does seem to occur in every humantissue type,indicating general importance in normal biological function (Figure 2AandB).In summary,the well-conserved eukaryotic nature,universal expression,generalized subcellular localization,and multidomain structure of GEMIN5 underscore its importance in normal biological function.Each of the three domains appears vital to healthy GEMIN5 function and the loss of a single one could have significant effects.Further,the G5C fragment and associated homodecamer exemplify the intricate nature of this largest member of the SMN complex.
GEMIN5 function
With regard to function,the role of GEMIN5 seems to vary based on its subcellular location and binding partner.As an SMN complex member,GEMIN5 serves as the pre-snRNΑ binding protein,fulfilling a critical role in successful snRNP biogenesis (Battle et al.,2006).This process appears to be dependent on the previously mentioned WD-40 repeat domains,which simultaneously interact with the m7G cap and Sm site of snRNA transcripts,ultimately allowing for the assembly of the Sm proteins around the Sm site by the SMN complex (Wahl and Fischer,2016).Further,GEMIN5 is also able to provide a level of quality control in this process,as it recognizes and rejects defective or unassembled presnRNΑ transcripts,guiding them to P-bodies for degradation,an action which likely explains the localization of the protein within these bodies (Fierro-Monti et al.,2006;Xu et al.,2016;Jiang et al.,2018).Αs a result,GEMIN5 provides a safeguard against defects in snRNA maturation and spliceosome assembly.Αs previously mentioned,GEMIN5 is also a direct binding partner of SMN via interaction between the noncanonical RBSs and the Tudor domain of SMN (Fortuna et al.,2023).Overall,GEMIN5 is an important member of the SMN complex,performing binding and regulatory functions important to accurate snRNP synthesis.
While the relationship between GEMIN5 and the SMN complex is clearly important,previously mentioned differences in localization patterns and observations of independence from the SMN complex emphasize the importance of understanding the SMN-independent functions of GEMIN5.In this state,GEMIN5 can be characterized as a translational regulatory protein as it possesses the ability to regulate mRNA expression and subsequent translation of bothSMN1and itself (Workman et al.,2015;Francisco-Velilla et al.2022a).Curiously,SMN also appears to be a positive regulator of GEMIN5 as SMN overexpression (OE) results in a significant increase in GEMIN5 levels within human iPSCs (Fortuna et al.,2023).In addition,given the presence of sequences similar to 3’ Sm sites within other mRNΑ transcripts,GEMIN5 may have further regulatory targets that have yet to be identified.With regard to the previously described G5C fragments,the proteolyzed GEMIN5 carboxyl terminus fragment also appears to possess the ability to bind to a variety of RNA targets depending upon secondary structure rather than nucleotide sequence (Pi?eiro et al.,2012;Guo et al.,2022).Αll these interactions further reinforce the role of GEMIN5 as a regulatory protein independent of the SMN complex.Furthermore,interactome analysis andin vitrodata has shown consistent associations with ribosomal proteins and polysomes with unique mRNA sequences,suggesting that GEMIN5 likely has further functions yet to be elucidated (Francisco-Velilla et al.,2016;Martinez-Salas et al.,2020;Embarc-Buh et al.,2022).Other known GEMIN5 interacting partners include GEMIN3,GEMIN4,CΑPRIN1,and eIF4E (Fierro-Monti et al.,2006;Battle et al.,2007;Vu et al.,2021;Delle Vedove et al.,2022).With this large variety of functions,it is easy to understand how dysfunction ofGEMIN5could yield pathologic results.
GEMIN5 syndrome
While the link betweenSMN1and SMA has been known for several decades,recent studies have begun to show that rare,pathogenic variants in other members of the SMN complex can manifest as multiple abnormal phenotypes distinct from SMA.Recently,multiple rare,pathogenic variants ofGEMIN5have been identified as the cause of a novel patient phenotype (Figure 3;Kour et al.,2021).While heterozygous individuals with a single copy of mutantGEMIN5do not show any obvious clinical symptoms,homozygous and compound heterozygous individuals display the previously mentioned unique NEDCΑM phenotype.This,along with the chromosomal location ofGEMIN5,suggests an autosomal recessive inheritance pattern (Kour et al.,2021;Saida et al.,2021;Rajan et al.,2022;Ibrahim et al.,2022;Zhang et al.,2023).
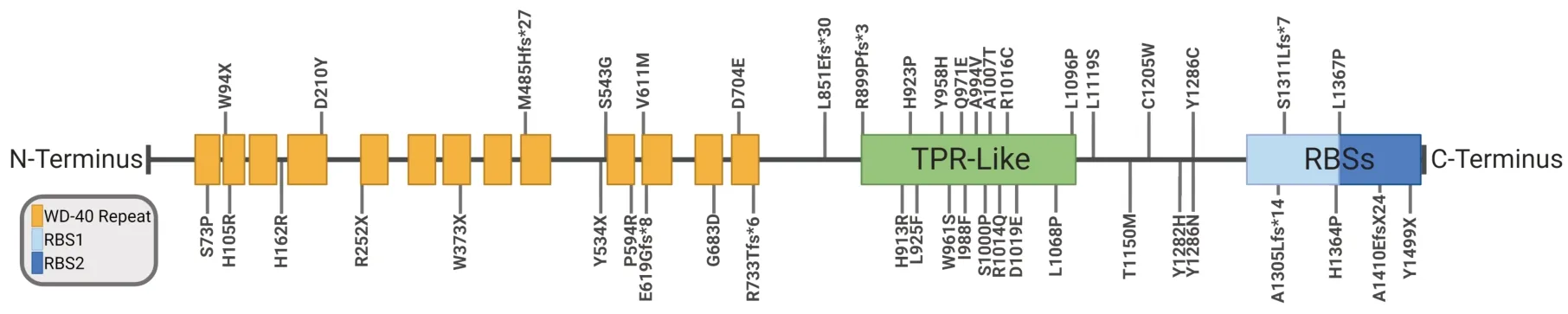
Figure 3| Disease-causing GEMIN5 variants have been identified in every domain of the protein.
As the name indicates,the most consistently observed signs and symptoms of NEDCAM are cerebellar atrophy,developmental delay,and motor dysfunction (Kour et al.,2021;Saida et al.,2021;Francisco-Velilla et al.,2022b;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).For most reported cases,the phenotype manifested itself within the first year of life;however,exceptions have been noted (Figure 4).Stratification of cases into distinct groups based upon age of onset and developmental delay,or characterization of motor dysfunction has been proposed but has yet to be formalized (Rajan et al.,2022).While the majority of cases appear to be static,progressive cases have been reported at a lower prevalence (Kour et al.,2021;Saida et al.,2021;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).Further,a variety of other common but not universally observed phenotypes have also been observed in NEDCAM patients,including,but not limited to,appendicular hypotonia,central hypotonia,absent or brisk deep tendon reflex,a progressive clinical course,infantile spasms syndrome with refractory epilepsy,and variable amounts of cognitive delay,all of which further broaden the clinical spectrum (Kour et al.,2021;Saida et al.,2021;Francisco-Velilla et al.,2022b;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).On the molecular level NEDCAM patient iPSC-derived neurons show distinct transcriptomic changes when compared with control.This trend continues when compared with SMA patient neurons,however,a slight overlap is present and,given the relationship betweenGEMIN5andSMN1,not unexpected (Kour et al.,2021).
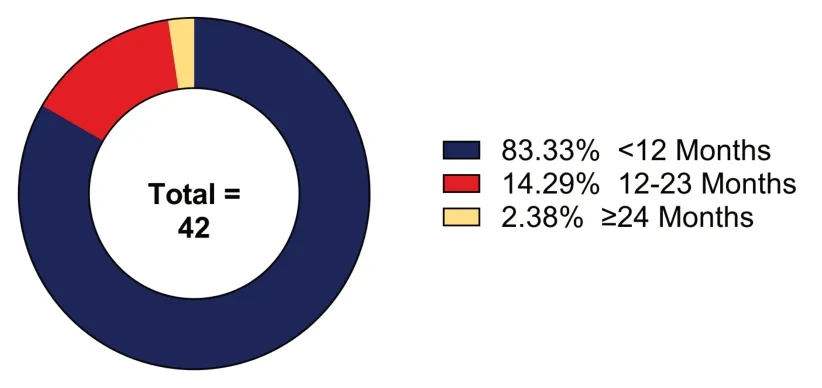
Figure 4| Stratified pie-chart of recorded ages of onset for GEMIN5 syndrome patients reported in the literature from Kour et al.(2021),Saida et al.(2021),Francisco-Velilla et al.(2022b),Ibrahim et al.(2022),Rajan et al.(2022),and Zhang et al.(2023).
NEDCAM patients overwhelmingly manifest signs and symptoms at an early age with a majority being observed within the first 12 months of life (Figure 4;Kour et al.,2021;Saida et al.,2021;Ibrahim et al.,2022;Rajan et al.,2022;Zhang et al.,2023).Given the multidomain,multifunctional nature of GEMIN5,it is possible that specific mutations are linked to certain observed phenotypes or ages of onset.A mutation perturbing the WD-40 repeats could potentially disrupt the ability of GEMIN5 to bind with snRNΑs but would have no effect on the functions of the RBSs.This potential genotype-phenotype correlation is at least partially supported by the observation that some mutations have a more severe effect on patients than others.For example,no patient homozygous for the p.His913Αrg mutation lived more than 3 months while two individuals bearing compound heterozygous p.Pro594Αrg/Ser543Gly mutations were both >55 years old at last observation (Kour et al.,2021;Rajan et al.,2022).While this example may indeed support the existence of this potential genotype-phenotype correlation,other factors,such as the effects of unknown modifiers or differences between the missense,nonsense,frameshift,and other mutations,could also be the cause.Thus,while all NEDCAM patients share a common clinical spectrum,further research and analysis is needed before any definitive conclusion can be drawn as to the source of the observed variations in disease severity and symptoms.
While the exact mechanisms by which NEDCAM manifests is not yet known,several theories exist to explain the mechanism of action.Currently,loss-of-function (LOF)mutations are thought to disrupt GEMIN5 function,resulting in NEDCAM pathogenesis (Kour et al.,2021;Saida et al.,2022;Fortuna et al.,2023).Disruption in expression level may also be a cause given the low level of GEMIN5 expression observed in patients,however,this would likely be linked with disruption in GEMIN5 cytoplasmic function as this decrease is not observed in nuclear GEMIN5 (Kour et al.,2021).Αs GEMIN5 is a multifunctional protein,there are several potential effects that mutations could have based on domain.Mutations within the WD-40 repeats could severely perturb snRNA binding and quality control,arguably the most critical function of GEMIN5 (Figure 5A).This disruption would,in turn,affect the entire function of the SMN complex and would likely result in decreased Sm core assembly around snRNA molecules (Figure 5B;Kour et al.,2021).This decrease would ultimately result in a reduction in snRNP expression,a result which may affect global spliceosome activity.This reduction could also theoretically occur within mutations located in the non-canonical RBSs,as the sites are responsible for protein-protein interaction between GEMIN5 and SMN(Fortuna et al.,2023),therefore affecting GEMIN5’s ability to participate in the SMN complex.Further,RBS1 mutations have been shown to reduce protein stability (Francisco-Velilla et al.,2022b).This mechanistic theory involving a decrease in spliceosomal activity is also reinforced by data showing that increased SMN expression can compensate for deficits in splicing machinery and defects in alternative splicing (Fortuna et al.,2023),lending further credence to this as the correct pathophysiology.

Figure 5| An illustration of normal GEMIN5 function both with and without the SMN complex.
While SMN complex disruption is currently thought to be the most probable cause of NEDCAM,mutations within the RBSs may have additional consequences as the domain is responsible for selective translation control.Disruption of this function could have a significant effect on protein expression,including that of SMN1 and GEMIN5 itself (Figure 5D;Workman et al.,2015;Francisco-Velilla et al.,2022a).Given the association ofSMN1with SMA,this may explain some of the shared transcriptional changes as well as shared symptoms,most notably the cerebellar atrophy observed in both GEMIN5 and SMΑ type III and IV patients as well as SMN?7 mice (de Borba et al.,2020;Kour et al.,2021;Cottam et al.,2023).
Regarding the TPR-like domain,evidence suggests that mutations within this domain reduce protein stability as well as disrupt normal GEMIN5 dimerization (Francisco-Velilla et al.,2022b).This disruption could then result in an increase in monomeric GEMIN5,though confirmation of this change is needed (Figure 5C).Further,mutations within the TPR-like and RBS1 domains have both been shown to disrupt proteinprotein interactions with TPR-like mutations decreasing both association and interaction with ribosomes (Francisco-Velilla et al.,2022b).Interestingly,TPR-like domain mutations have been associated with some of the less common NEDCAM symptoms,such as intellectual disability and infantile spasms,however the reason for this is not clear (Ibrahim et al.,2023;Zhang et al.,2023).
Current Animal Models
As previously mentioned,SMN complex proteins are important regulators of a fundamental cellular process,thus,it is not surprising that many SMN complex proteins are evolutionary conserved at functional and structural levels between species.Interestingly,the complexity of the SMN complex increases with evolution and may acquire additional functions,thus,the use of eukaryotic models is most beneficial within this area of study (Pi?eiro et al.,2015).Similarly,while several cell lines,including patientderived iPSCs have been used to study the effects of mutant GEMIN5 at the cellular level (Kour et al.,2021;Fortuna et al.,2023),whole organism genetic models are also necessary to understand the complete pathogenesis and pathophysiology of NEDCAM.Thus,common eukaryotic model organisms within the field of genetics,such as fruit flies (Drosophila melanogaster),zebrafish (Danio rerio),and mice (Mus musculus) have been utilized in the study ofGEMIN5.
Drosophila
Close to 75% of known disease-causing genes found in humans are also found in the fruit fly genome (Reiter et al.,2001).Further,given that fruit flies have previously been used to develop models of other neurodevelopmental diseases (Kim et al.,2020),the use ofDrosophilain investigatingGEMIN5mutations seems appropriate.While theDrosophilaSMN complex is significantly simpler than the complex found within vertebrates (Kroiss et al.,2008),it does possess a GEMIN5 ortholog referred to asrigor mortis(rig) (Gates et al.,2004).The encoded rig protein possesses~73% (MUSCLE) identity to human GEMIN5 and was initially identified as a nuclear receptor protein within the ecdysone signaling pathway (Gates et al.,2004;Cauchi et al.,2010;Madeira et al.,2022).Notably,rigLOF was reported to be associated with a motor phenotype several years prior to the first mention of aGEMIN5patient within the literature (Borg and Cauchi,2013,2014).Further,globally enhancedrigknockdown (KD) shows significant larval lethality,indicating a critical role in developmental processes(Borg and Cauchi,2013).Rig possesses WD-40 repeats but has a substantially different carboxyl terminus possessing a LXXLL motif (Gates et al.,2004).Despite this structural difference,evidence suggests rig is indeed a member of the SMN complex inDrosophila,though it should be noted that the complex lacks orthologs for GEMINs 6–8 and thus may have altered functions due to fundamental differences between the invertebrate and vertebrate nervous systems (Matera et al.,2019).
Given the significant difference in structure between GEMIN5 and rig,mutations imitating those observed in patients could not be induced.As a result,the current fruit fly model uses inducible RNAi KD via the UAS-Gal4 system to reduce rig expression levels.This system is then able to mimic the suspected LOF modality responsible for NEDCAM in patients.The model displays complete pupal lethality,indicative of significant late-stage developmental defects,as well as significantly reduced larval bouton size.Further,whenrigKD was induced in adult flies,significant reductions in climbing ability and significant increases in mortality rate were observed when compared to control (Kour et al.,2021).Recently,this model was also used to conduct a genetic screen for modifiers ofrig,leading to the evaluation of possible compensatory effects due to up-or downregulation of specific interacting partners.WhileGemin2upregulation resulted in an exacerbation in phenotype,Smnupregulation via OE resulted in significant phenotype suppression when compared to control.This suppression was non-sex dependent and perpetual as flies aged (Fortuna et al.,2023).
Overall,theDrosophilamodel has provided significant insights into the role ofGEMIN5in neurodevelopmental disorders.The model effectively mimics a number of observable markers within the human phenotype,including the reduction in protein levels,developmental defects,and impaired motor function,reinforcing the importance of GEMIN5 and its orthologs for normal organismal development and motor function.While other animal models with greater genetic similarity to humans have been developed,theDrosophilamodel still has utility.Further use in genetic screening studies to identify additional modifiers ofrigexpression,as well as further refinements of the model to mimic the human phenotype more closely are two potential future uses.
Zebrafish
Zebrafish models have also been used to studyGEMIN5.The zebrafish ortholog of GEMIN5 shares only a 51% identity to the human protein (Saida et al.,2021),yet the overall structure of the two polypeptides is remarkably similar,each possessing clear WD-40 repeat,TPR-like,and noncanonical RBS domains.Still,the two proteins have significant differences which must be accounted for when creating a model.Four zebrafish models have been created thus far,all of which result in the expression of a truncated form of gemin5.Notably,while two of these models were created using CRISPR/Cas9 with the specific intent to mimic known GEMIN5 mutations in humans (Saida et al.,2021),two were created in a random mutagenesis screen (Pei et al.,2020;Liu et al,2021),potentially limiting its translational utility in human-focused research.Zebrafish expressing mutantgemin5in a homozygous manner show clear developmental defects as larva,including decreased cerebellum size,shorter body length,smaller heads with unusual eyes,fewer hair cells,compromised regenerative ability in select tissues,and impaired definitive hematopoiesis (Pei et al.,2020;Liu et al.,2021;Saida et al.,2021).Furthermore,mutantgemin5zebrafish had significantly different survival rates when compared with control,and no homozygotes survived more than 12 days after hatching (Saida et al.,2021).Notably,while the truncation of the protein occurs at varying locations in the different zebrafish lines,all variants with abnormal phenotypes lost at least part of the RBS domains,indicating the importance of the domain for proper development.This observation may also be applicable to human patients due to the previously mentioned highly conserved residues responsible for RNA binding activity;however,it is not clear if this domain also facilitates binding with the zebrafish ortholog ofSMNas is observed in humans (Fortuna et al.,2023).Interestingly,the reduction in hair cells,compromised regenerative ability,and defective hematopoiesis observed in zebrafish mutants has not been observed in humanGEMIN5patients,though it may be beneficial to re-examine known patients for these issues.Overall,zebrafish prove to be an effective model organism for the study of NEDCAM and have thus far emphasized the importance of the RBS1 and RBS2 domains.Further,the defects observed in larval zebrafish homozygous for pathogenicgemin5variants provide additional support for the connection betweenGEMIN5and cerebellar volume loss in humans.However,the notable genetic differences between zebrafish and humans may limit the model’s applicability and utility,thus,a model which more closely reflects the human condition is needed.
Mice
To date,only one study has used mice to investigateGEMIN5mediated neurodevelopmental disorder.Laboratory mice are an excellent candidate for use in the creation of a NEDCAM model for a number of reasons.Mice are a relatively small mammal with a high level of fecundity and a short generation time,factors which allow many animals and multiple generations to be studied by a single researcher (Craigen,2019a).Further,mice come with the additional benefits of being extremely well characterized,having the ability to engage in complex behaviors,and possessing a high degree of genetic similarity to humans (Craigen,2019a).With regard to investigatingGEMIN5,mice are an excellent choice given the high degree of similarity between the human protein and its mouse ortholog,Gemin5.The two genes are over 85%identical (BLASTn) (Sayers et al.,2022) and the expressed proteins only differ in length by five to seven residues depending upon the transcript (Table 1).

Table 1|The chromosomal locations and molecular weight of SMN complex members in both humans and mice
In the aforementioned study,Gemin5knockout (KO) mice were generated via CRISPR/Cas9 in a C57Bl/6NJ background strain.Similar to humans,heterozygous KO mice had no observable phenotype;however,homozygous KO pups were never observed (Rajan et al.,2022).These observations suggest complete embryonic lethality and were further investigated by evaluating the genotypes of embryonic tissue between E9.5 and E10.5.No homozygous embryos were detected at this stage,meaning that the embryos were non-viable prior to thistimepoint.This suggests thatGemin5may play a critical role during the initial stages of mammalian development,however,given the lack of homozygous embryos and animals,the effect on development,particularly in the brain,is still unknown(Rajan et al.,2022).
Tangentially,SMN?7 mice used in the study of SMΑ show significant cerebellar alterations including volume loss as well as functional abnormalities contributing to a motor phenotype(Tharaneetharan et al.,2021;Cottam et al.,2023).While this further reinforces observed similarities between NEDCAM and SMA symptoms,it also suggests that a mouse model of NEDCAM with cerebellar atrophy may be possible to develop.Overall,laboratory mice are well situated for further use to investigate GEMIN5 due to their genetic similarity to humans as well their complex and well-characterized nature.The KO Gemin5 mice once again emphasize the crucial role of the protein in development.Future investigations using mice have the potential to elucidate the function of GEMIN5 in development,especially within the brain,and to increase the understanding of NEDCAM pathogenesis.
Modifiers and Potential Therapies
Αs no treatment currently exists for GEMIN5 syndrome,clinicians have been focused primarily on symptoms management.This palliative approach includes a variety of medications to alleviate symptoms as well as physical therapy to address motor dysfunction.While this may be effective for improving quality of life,the root cause of NEDCAM is not being addressed.Thus,the use of animal models to increase understanding of GEMIN5 presumed partial LOF variants and NEDCΑM pathophysiology,as well as to identify possible therapeutic targets,is critical.
To date,all animal models ofGEMIN5have shown the importance of the protein with regards to proper development.While bothrig/gemin5LOF Drosophila and zebrafish larva are viable,their survival rate decreases over time,ultimately resulting in complete larval lethality (Kour et al.,2021;Saida et al.,2021).This trend continues within mice,as no homozygousGemin5KO pups are able to survive past the early stages of embryonic development (Rajan et al.,2022).While the miscarriage rate due to NEDCAM is unknown and poses one of many future directions of study,it appears that the survival of any NEDCAM patients further into development is possibly unique to humans.This may be due to the partial LOF nature of most of the mutations and given that significant variations in disease course and onset have been observed between patients,it is likely that a genotype-phenotype correlation or a number of genetic modifiers may also be involved in NEDCAM manifestation.In particular,the effects of genetic modifiers are of unique interest given their effect on SMΑ.For instance,a variant of theHspa8gene has been shown to alleviate SMA symptoms within a humanized mouse model (Kim et al.,2023).Further,modifications in the expression levels of various proteins,such as the downregulation of members of the transcription export complex or the upregulation ofZpr1have both been shown to result in the upregulation ofSmn1within HEK cells and SMA mice,respectively (Kannan et al.,2020;McCormack et al.,2021).Given the clear link between this disease and NEDCAM,it is logical that modifiers ofGEMIN5can be identified.
SMN has recently been identified as one of these modifiers and may provide an eventual path to treatment.The relationship between GEMIN5 and SMN is very strong as the proteins are direct binding partners and appear to influence each other’s expression.This connection has also been the source of previous speculation thatGEMIN5may act as a modifier of SMΑ,along with other GEMIN proteins (Borg and Cauchi,2014).As previously mentioned,SMNOE provides significant compensatory effects withinrigKD flies when compared with control,indicating that SMN is a significant modifier (Fortuna et al.,2023).This compensatory effect has also been shown to occur within a number of cell typesin vitro.HEK293T and iPSC-derived GEMIN5 patient neurons withSMN1OE both show a significant increase in expressed GEMIN5.Though the specific mechanism behind this trend remains unclear,it is not entirely unexpected given the aforementioned relationship between the two proteins.Furthermore,previous data has confirmed the presence of decreased SMN1 levels within GEMIN5 patients while more recent data has confirmed the presence of decreased GEMIN5 levels within SMA type I and II patient motor neurons (Kour et al.,2021;Fortuna et al.,2023).Though the application of SMN upregulation for treatment may not work on all known pathogenic variants,the positive relationship emphasizes the importance of GEMIN5 and SMN’s connection.Further,given the unique presence ofSMN2in humans,the relationship between the proteins may potentially explain survival into later developmental stages.AsSMN2does produce functional RNΑ transcripts,albeit at a significantly lower rate thanSMN1,this may result in an increase in overall SMN levels,however,further investigation into this topic is needed before this conclusion can be drawn.Continuing forward,the positive correlation in expression between the two proteins can also be observed when both cell types are treated with the FDΑ-approved SMΑ antisense oligonucleotide Nusinersen (Fortuna et al.,2023).Αs previously mentioned,NEDCAM patients currently resort to palliative treatments,thus,potential therapeutic pathways,especially those with existing medication,warrant attention.Observed increases in GEMIN5 expression within human cell lines after Nusinersen administration are an especially important development as it provides the first potential treatment path for NEDCAM patients.Increased SMN expression also shows significant downstream effects such as decreases in alternative splicing defects and increases in the presence of spliceosome components,such as snRNPs,within Cajal bodies (Fortuna et al.,2023).Αs previously mentioned,this increase lends credence to the theory that spliceosome deficits are the primary cause of NEDCΑM;however,further research is still needed for definitive confirmation.Further,given the recent FDA approval of the orally administered SMA medication Risdiplam,there may already be multiple therapies available for use in treating NEDCΑM.
Conclusion and Future Directions
IdentifyingGEMIN 5as the cause of a novel neurodevelopmental disorder highlights the importance of members of the SMN complex in healthy human development.Along withGEMIN5andSMN,GEMIN4has also recently been shown to cause another unique disease phenotype (Aldhalaan et al.,2021).This not only emphasizes the importance of these proteins in maintaining biological norms but also strongly reinforces previous theories that the dysfunction of other SMN complex members could result in novel phenotypes (Borg and Cauchi,2014;Pei et al.,2020).Thus,renewed investigation into these proteins is well warranted.
In recent years,understanding of the function of GEMIN5 has significantly increased.The scope of research into this gene has been widened substantially and many important discoveries regarding function,structure,localization,and mechanism have been made within the past 5 years alone.While NEDCAM currently has no treatment or cure,understanding of the disease has increased rapidly and a potentially viable treatment pathway has been identified.SMN upregulating therapies have previously been developed for the treatment of SMA.Although current data suggests that these therapies have the potential to increaseGEMIN5expression and treat NEDCΑM (Fortuna et al.,2023),further work must be done to validate these findings on the organismal level before they can be considered for use in treatment.
While the use of SMN upregulation therapies is promising,there is still much more to learn about GEMIN5 disorder and its various manifestations.Determining the potential effects of specific mutations in each domain as well as determining how the mutations influence symptoms may be important to understand the clinical spectrum currently observed in patients,as well as to understand the role of other potential modifiers.As previously mentioned,comprehending why patients appear more viable than any other species is also of interest,as it may lead to greater cognizance of NEDCAM specifically and the functions of the SMN complex in general.Further,knowledge regarding the natural progression history of NEDCAM is also lacking as many patients have only been under observation for <5 years.Thus,pediatric follow-up is needed in the coming years using a battery of radiological and neurological techniques.Questions remain regarding the nature of the cerebellar volume loss observed within patients as well as the pathophysiology behind this manifestation.This is also related to the current uncertain clinical course of NEDCΑM.Αs previously mentioned,most cases appear to be static,though at least four cases in the original report noted a progressive clinical course (Kour et al.,2021).Despite this,the short term of observation for most of these patients should be taken into account as while the course may currently appear static,it may be deemed progressive in the long term.
Regardless of the path that future investigation follows,the study ofGEMIN5clearly needs a more accurate animal model.Αs previously mentioned,the use of mice to facilitate investigations intoGEMIN5has great potential due to the many benefits of the model as well as the great degree of similarity between the orthologous proteins.While mice bearingGemin5mutations with a high degree of similarity to those reported in humans could easily be engineered via CRISPR/Cas9,it is possible that the embryonic lethality issues observed within the KO mice could also occur.This concern is reinforced by the larval lethality observed in both theDrosophilaand zebrafish models (Kour et al.,2021;Saida et al.,2021).Potential solutions include mutagenesis via AAVs during gestation or post-parturition as well as the creation of a conditional model via the Cre-LoxP system,both of which have been used in the study of neurodevelopmental diseases in the past (Craigen,2019b;Kao et al.,2020).The Cre-LoxP system in particular would provide a versatile platform for the creation of a mouse model as it is highly developed,very well characterized,and would provide high levels of spatiotemporal control.The use of a conditional model for the study of GEMIN5 has already been shown to be feasible through the use of the inducible UAS-GAL4 system within Drosophila (Kour et al.,2021;Fortuna et al.,2023).Further,the existence of SMN upregulation therapy reactive mouse models previously used in the study of SMA (Berciano et al.,2020) could potentially be crossed with mutant GEMIN5 mice.The resulting hybrid mouse lines could then be used to test SMN upregulation therapies,providing valuable insight into their use as a potential treatment for NEDCΑM.
In summary,due to its novelty,many questions about GEMIN5 syndrome still remain;however,given the speed of recent advances and the recent rise in interest,it is likely that many of these questions will be answered in the coming years,leading to an increased understanding and,hopefully,the development of a treatment for this novel neurodevelopmental disorder.
Acknowledgments:The authors are thankful to Dr.John R.Chaillet and Madeleine Lorenger for assistance in editing and to Dr.Sukhleen Kour for assistance with figures.
Author contributions:The manuscript was written by CHN and revised by UBP.Both authors read and approved the final manuscript for publication.
Conflicts of interest:The authors declare that there is no conflict of interest regarding the publication of this paper.
Data availability statement:The data are available from the corresponding author on reasonable request.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Encarna Martinez Salas,Universidad Autonoma de Madrid,Spain;Jianli Sun,Delaware State University,USA.
Additional file:Open peer review reports 1,2.

- 中國神經再生研究(英文版)的其它文章
- A sphingolipid message promotes neuronal health across generations
- Krüppel-like factor 2 (KLF2),a potential target for neuroregeneration
- Defined hydrogels for spinal cord organoids: challenges and potential applications
- Neuronal trafficking as a key to functional recovery in immunemediated neuropathies
- Advancements in personalized stem cell models for aging-related neurodegenerative disorders
- New insights into astrocyte diversity from the lens of transcriptional regulation and their implications for neurodegenerative disease treatments